Šta je talasemija?
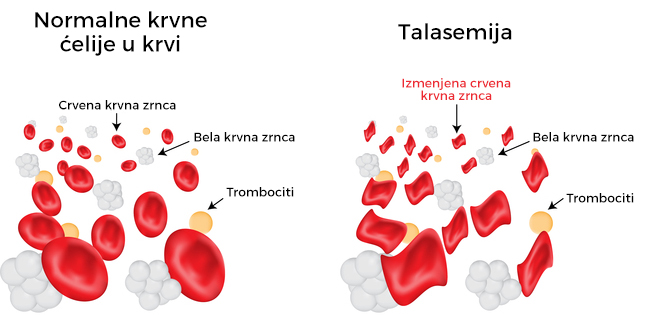
Talasemija predstavlja nasledni poremećaj krvi koji nastaje zbog nepravilnog stvaranja jednog od četiri lanaca aminokiselina koje čine hemoglobin.
Talasemija, zajedno sa anemijom srpastih ćelija je među dominantnim bolestima u zemljama u razvoju, u kojima se zbog malih mogućnosti prenatalne dijagnostike održala vrlo visoka stopa incidence ovih naslednih oboljenja.
Naziv talasemija vodi poreklo od grčkih reči Thalassa (more) i haima (krv). Kada je otkrivena, smatralo se da je bolest raširena isključivo među stanovnicima Sredozemlja, te je nazvana i Mediteranska groznica. Danas se, zbog masovnih migracija stanovništva, ovaj poremećaj prepoznaje u različitim delovima sveta.
Talasemije se ubrajaju u nasledna oboljenja, a glavno obeležje je postojanje poremećaja u stvaranju globina, koji predstavlja dio hemoglobina, proteina koji je odgovoran za dopremanje kiseonika svim ćelijama i tkivima čovjekovog organizma. Talasemije nastaju kao posledica različitih defekata na nivou gena.
Neke talasemije do kraja života ostaju asimptomatske, ali su češće one koje mogu da prouzrokuju skraćen životni vek eritrocita i hemoliznu anemiju, tj. razaranje crvenih krvnih zrnaca. Svaka od navedenih manifestacija prouzrokuje dalje patološke procese.
Određene vrste talasemija su češće u određenim delovima sveta.
Talasemije uglavnom pogađaju oba pola podjednako.
Podela talasemija
Zavisno od prisutnih genetskih promena, postoji više varijanti bolesti.
Na osnovu toga koji globinski lanac hemoglobina je prisutan u manjku, razlikuju se dva oblika talasemije:
- Alfa talasemija
- beta talasemija
Talasemije su nasledna monogenska oboljenja, što znači da su uslovljene mutacijom (promenom) jednog gena. Promena u genu odgovornom za nastanak talasemije može biti prisutna samo kod jednog ili kod oba roditelja.
Podela alfa i beta talasemija
Zavisno od toga da li su oba roditelja nosioci mutiranog gena ili nisu, i alfa i beta talasemija se mogu podeliti na:
Talasemija major
Talasemija major – najteži oblik talasemije, javljaju se simptomi teške anemije, nastaje kod osoba kod kojih su oba roditelja nosioci mutiranog gena. Za takve osobe kažemo da su homozigoti za talasemiju.
Talasemija intermedia
Talasemija intermedia – srednje težak poremećaj, anemija je blaga do umerena, javlja se kod osoba kod kojih je samo jedan od roditelja nosilac mutiranog gena. Za osobe kod kojih postoji talasemija intermedia kaže se da su heterozigoti za talasemiju.
Talasemija minor
Talasemija minor – osobe su uglavnom asimptomatske ili se ispoljavaju vrlo blagi simptomi, nastaje ukoliko je samo jedan od roditelja nosilac defektnog gena i ove osobe su takođe heterozigoti za talasemiju.
Šta je uzrok talasemija?
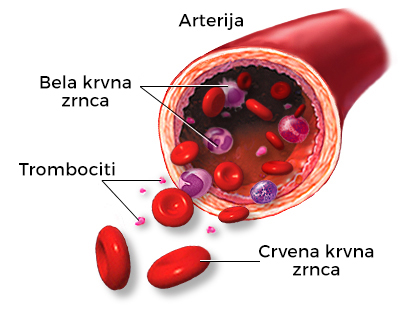
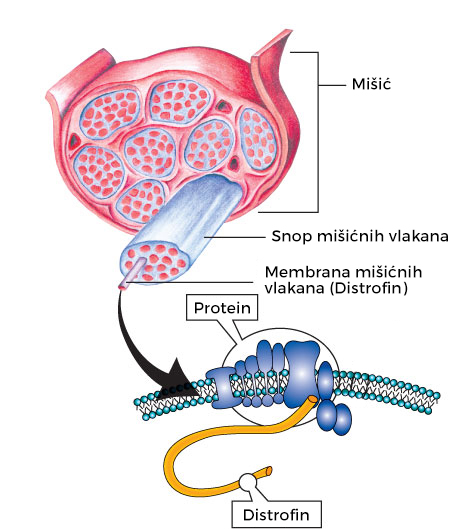
Kako bi se bolje razumeo uzrok nastanka talasemija, korisno je znati strukturu hemoglobina, proteina koji je zadužen za transport kiseonika od pluća do svih ćelija u organizmu. Hemoglobin se nalazi u crvenim krvnim zrncima (eritrocitima).
Od čega se sastoji hemoglobin?
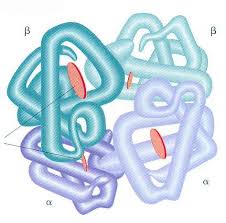
Hemoglobin se sastoji iz tri različite funkcionalne i strukturne jedinice:
1. Hem
2. Globin – proteinski lanci
3. Atom gvožđa – nalazi se u središtu hema
Globin je izgrađen od po 2 identična para polipeptidnih lanaca i ti parovi se obeležavaju grčkim slovima – α2, β2, γ2, δ2.
Tipovi hemoglobina
Na osnovu kombinacije parova ovih lanaca, razlikuju se četiri tipa hemoglobina:
1. Fetalni hemoglobin (HbF) – α2γ2
2. Adultni hemoglobin (HbA) – α2β2
3. Adultni 2 hemoglobin (HbA2) – α2δ2
4. Primitivni (embrionalni) hemoglobin (HbP) – α2ε2
Koji će tip hemoglobina u organizmu biti prisutan zavisi od životnog doba. Tako će u fetalnom periodu života zbog specifičnih uslova u materici majke u bebinim eritrocitima biti 100% prisutan fetalni hemoglobin HbF. Njegov procenat se postepeno smanjuje – pri rođenju iznosi 60-80%, da bi u petom mesecu pao na 10%. Ostalih 90% od ukupnog hemoglobina u krvi biva zamenjen adultnim hemoglobinom (HbA).
Hemoglobin A je glavni hemoglobin kod odraslih i sadrži dva alfa globin lanca. Kod zdravih odraslih osoba je prisutan i adultni 2 hemoglobin (HbA2) – α2δ2.
Sinteza globinskih lanaca hemoglobina je genski predodređena, tako se geni za sintezu alfa (α) HBA1 i HBA2 lanca nalaze na hromozomu 16, a gen HBB za beta lanac na hromozomu 11.
Ukoliko dođe do promene (mutacije) u ovim genima, izmenjeni geni nastavljaju da kodiraju sintezu fetalnog hemoglobina i nakon porođaja. Kao posledica ovoga, dobija se velika količina fetalnog hemoglobina u krvi odrasle osobe koji bi u normalnim uslovima trebao biti odsutan. Na ovaj način nastaje posebna grupa poremećaja koja se jednim imenom zovu talasemijski sindromi.
Alfa talasemija
Alfa (α) talasemija je češća u jugoistočnoj Aziji, Indiji, Bliskom Istoku i Africi. Javlja se kod oba pola sa podjedankom učestalošću, s tim što alfa talasemija sa mentalnom retardacijom pogađa isključivo pripadnike muškog pola.
Alfa talasemija je nasledna, urodjena bolest, te se simptomi i znaci javljaju kod dece i novorođenčadi.
Šta je uzrok alfa talasemije?
Alfa talasemija se javlja kada dođe do defekata u dva gena za alfa globin. Kod alfa talasemije, sinteza alfa lanaca je smanjena ili u potpunosti nedostaje, što dovodi do smanjene količine adultnog hemoglobina.
Ispoljavanje i težina alfa talasemije će zavisiti od broja mutacija u genima HBA1 i HBA2.
Smatra se da je jedna od 25 osoba nosilac mutacija u HBA1 ili HBA2 genu.
Koji su simptomi alfa talasemije?
- Skraćenje životnog veka eritrocita
- Snižen nivo gvožđa
- Mikrocitoza – crvena krvna zrnca smanjene veličine
- Povećanje broja crvenih krvnih zrnaca -eritrocitoza
- Žutica kod novorođenčadi
- Anemija
- Uvećanje jetre
- Uvećanje slezine
- Kamenci u žuči
- Smanjenje gustine kostiju
- Sklonost ka prelomima kostiju
- Izražena gornja vilica
- Niži rast
Kako se nasleđuje alfa talasemija?
Talasemija major se nasleđuje autozomno recesivno. Ovaj tip nasleđivanja podrazumeva da je za ispoljavanje bolesti kod deteta, potrebno da oba roditelja da budu nosioci mutacije u HBA1 ili HBA2 genu. Rizik da dete ima alfa talasemiju zavisi od toga da li su roditelji nosioci bolesti ili ne.
Danas postoje genetički testovi za tzv. skrining nosilaca bolesti kojima se, još pre začeća, može utvrditi da li su roditelji nosioci mutiranih gena, kako za alfa, tako i za beta talasemiju, ali i za brojna druga nasledna oboljenja. Ovi testovi se takođe mogu uraditi i tokom trudnoće, ili po rođenju deteta.
Osoba koja je nasledila jedan mutirani gen od jednog od roditelja neće pokazivati simptome alfa talasemije.
Osoba sa dva mutirana gena ispoljava samo umerenu mikrocitozu (smanjena veličina crvenih krvnih zrnaca) i hipohromiju (slaba obojenost eritrocita, zbog manjka hemoglobina). Kod osoba sa dva mutirana gena nisu prisutne anemija i hemoliza (raspadanje crvenih krvnih zrnaca).
Ukoliko postoji defekt u tri gena za alfa lanac globina, izražene su hemoliza, hipohromija i veliki broj eritroicita u perifernoj krvi koji imaju izgled mete (target ćelije).
Ukoliko dođe do defekta u sva četiri gena alfa globin, razvija se stanje koje se naziva fetalni hidrops. Ovo stanje je ranije bilo inkompatibilno sa životom. Danas, ukoliko je rizik unapred poznat, intrauteralna transfuzija (transfuzija fetusa dok se još nalazi u materici) može omogućiti normalnu trudnoću. Ovakva deca zahtevaju hroničnu transfuziju nakon porođaja i često, transplantaciju koštane srži.
Hemoglobin H Constant Spring je varijanta alfa talasemije u kojoj postoje defekti dva gena alfa globin plus jedan alpha globin gen je mutiran.
Kako se postavlja dijagnoza alfa talasemije?
Kod sumnje na alfa talasemiju, prvo se radi analiza kompletne krvne slike. Može se uraditi laboratorijski pregled razmaza periferne krvi, kao i elektroforeza hemoglobina.
Zlatni standard u dijagnostici alfa talasemije je genetičko testiranje. Genetičkim testiranjem se zapravo vrši analiza delecija/duplikacija HBA1 i HBA2 gena i detekcija HbCS (Hemoglobin Constant Spring) mutacije.
Kako se leči alfa talasemija?
Osobama sa alfa talasemijom propisuju se preparati folne kiseline, uz izbjegavanje suplemenata koji sadrže gvožđe. Gvožđe se može taložiti u jetri i dovesti do insuficijencije jetre. Kod dece kod kojih je izvršeno hirurško otklanjanje slezine, koriste se antibiotici u cilju prevencije različitih bakterijskih infekcija. Transfuzija krvi se preporučuje samo u hitnim slučajevima, ne redovno, kako ne bi došlo do nakupljanja gvožđa. Pacijenti sa varijantom Hemoglobin Constant Spring mogu imati značajnu anemiju i zahtevaju česte transfuzije tokom života.
Kod sumnje na alfa talasemiju, radi precizne dijagnoze, najpouzdanije je sprovesti genetičko testiranje, kako bi se precizno utvrdio koji tip talasemije je prisutan, jer od toga zavisi kako lečenje, tako i prognoza. Kod osoba koje su nosioci mutiranih gena, preporučuje se prenatalno testiranje u cilju utvrđivanja rizika postojanja alfa talasemije kod potomstva.
Beta talasemija
Beta (β) talasemija je mnogo češća u mediteranskim zemljama, kao što su Grčka, Italija, Španija, zatim mediteranskim ostrvima kao što su Kipar, Sardinija i Malta. Beta talasemija je takođe češća u Istočnoj Evropi nego u drugim delovima sveta.
Nastaje usled promjena na nivou HBB gena koji se nalazi na 11-om paru hromozoma, a odgovoran je za stvaranje beta lanaca globina.
Kod beta talasemije je često prisutna anemija koja se može manifestovati lupanjem srca, bledilom lica, hroničnim umorom i malaksalošću. Zbog povećanog nivoa bilirubina može biti prisutna žutica. Kod nekih osoba dolazi do pojave aritmija, a kod nekih se javlja uvećanje jetre i kamenci u žučnoj kesi koji se obično registruje nakon što osoba oseti nelagodnost u desnoj strani trbuha, ispod rebara.
Simptomi se mogu javiti u detinjstvu, ali i kasnije tokom života, što može otežati dijagnostiku.
Kao i za alfa talasemiju, i za beta oblik talasemije danas postoje genetički testovi kojima se može kako pre, tako i po rođenju deteta utvrditi da li je dete nosilac mutacije u HBB genu, odnosno da li ima rizik za razvoj bolesti. Takođe, moguće je da parovi koji planiraju potomstvo, još pre začeća utvrde da li su nosioci mutacije u HBB genu, odnosno, da li će njihova deca imati rizik za beta talasemiju.
U tu svrhu, naša preporuka je da poverenje poklonite genetičkom skrining testu Adventia. Jednostavno testiranje podrazumjeva uzimanje bukalnog brisa roditelja koji se prosleđuje na dalju analizu. Zahvaljujući savremenim metodama danas je moguće iz samo jednog uzorka bukalnog brisa analizirati veliki broj klinički relevantnih gena, što je u poslednjih par godina veliki napredak. Iskoristite napredak tehnologije u korist saznanja da li ste potencijalni prenosnig mutiranog gena koji potomstvu može izazvati razvoj nekog od oblika talesemije.