PremiumGene - Najobuhvatniji i Najdetaljniji Prenatalni Test u Srbiji
PremiumGene i PremiumGene + SMA su najobuhvatniji i najdetaljniji prenatalni testovi koji istovremeno analiziraju aneuploidije, mikrodelecione sindrome i 100 monogenskih bolesti uz mogućnost otkrivanja statusa nosilaca za spinalnu mišićnu atrofiju (SMA) i pol bebe.

Najčešće trizomije
+ mikrodelecije
+ 100 monogenskih bolesti
+ pol
još pre rođenja bebe

Šta je
PremiumGene test?
Neinvazivni prenatalni test (NIPT)je skrining test koji već od 10. nedelje trudnoće može detektovati postojanje najčešćih hromozomskih abnormalnosti ploda. Testom se analizira slobodnocirkulišuća DNK bebe u krvi majke, radi otkrivanja izuzetno teških genetičkih poremećaja u ranoj trudnoći. Neinvazivni prenatalni test (NIPT) je potpuno bezbedan i precizan, te predstavlja odličan način da se pre rođenja bebe utvrdi postojanje rizika na pojavu određenih genetskih poremećaja.
PremiumGene je prvi sveobuhvatni neinvazivni prenatalni test koji može istovremeno da analizira aneuploidije hromozoma, mikrodelecije, a dodatno još i 100 monogenskih bolesti i pol bebe.
PremiumGene je precizan i potpuno bezbedan neinvazivni prenatalni test (NIPT), dostupan kod jednoplodnih i blizanačkih trudnoća, kao i kod trudnoća koje su začete vantelesnom oplodnjom.
Može se uraditi već od 10. nedelje trudnoće. Test je bezbedan i za majku i za bebu, jer je za njegovo izvođenje potreban samo uzorak venske krvi majke i bukalni bris biološkog oca.
PremiumGene i PremiumGene + SMA predstavljaju apsolutno najobuhvatnije prenatalne testove na svetu, jer sa dodatnom analizom monogenskih bolesti, pružaju apsolutno najdublji pogled u genetsko zdravlje bebe u najranijoj fazi trudnoće.

Jedini prenatalni test u kojem učestvuju i tate!




Kako se radi Premium Genetics test?
Samo izvođenje testa je veoma jednostavno. Potrebno je da pozovete naš stručni tim radi više informacija i zakazivanja uzorkovanja,
na broj 064 659 24 28. Samo uzorkovanje možete obaviti u bilo kom gradu u Srbiji, u nekoj od naših partnerskih laboratorija koja je dostupna u vašem mestu stanovanja.
Za PremiumGene test potreban je:
- Uzorak krvi majke
- Bukalni bris biološkog oca
* dodatni uzorak bukalnog brisa neophodan je za panele koji uključuju i SMA

Nakon završenog uzorkovanja, Premium Genetics zakazuje kurirsku službu, koja preuzima Vaš uzorak. DHL je dalje zadužen za transport uzorka u inostranu laboratoriju. Uzorak u laboratoriju stiže najčešće u roku od 24 do 48 sati, u specijalnom setu sa kriogelom, koji garantuje stabilnost uzorka.

Premium Genetics prenatalni testovi koriste novu tehnologiju obogaćenog targetiranja, koja omogućava superiornu detekciju aneuploidija i mikrodelecija uz neprevaziđenu tačnost merenja fetalne frakcije (udeo bebine DNK u krvi majke). Ciljani regioni na hromozomima 21, 18, 13, X i Y su locirani, zumirani i analizirani na aneuploidije, koristeći naše sopstvene genetičke i bioinformatičke tehnologije. Superiornost analize se ogleda u izveštavanju o triploidijama uz mogućnosti analize od 10. nedelje trudnoće.
Superiornost PremiumGene testa ogleda se u mogućnost analize mikrodelecionih sindroma i kod blizanačkih trudnoća, što je ograničenje ostalih, starijih generacija neinvazivnih prenatalni testova, koji su prisutni kod nas.
Zašto izabrati Premium Genetics test?
Postoji mnogo razloga zbog kojih bi budući roditelji trebali da se odluče baš za Premium Genetics test sa monogenskim bolestima. Pre svega, superiornost tehnologije koja omogućava analizu na nivou jednog gena, odnosno mogenskih bolesti, sama po sebi dokazuje razliku u odnosu na druge prenatalne testove.
PremiumGene i PremiumGene + SMA su apsolutno najobuhvatniji prenatalni testovi na svetu! Posebno je važna analiza monogenskih bolesti, jer za ovu vrstu bolesti ne postoje biohemijski markeri, niti su vezani sa standardnim faktorima rizika. Svako može biti nosilac monogenske bolesti, a da nema simptome, niti da je toga svestan. Upravo zato je ova informacija od velikog značaja i zbog toga je Premium Genetics test najsuperiorniji neinvazivni prenatalni test na svetu što daje budućim roditeljima mogućnost da na vreme donesu pravu odluku.
Ukoliko imate bilo kakve dileme, možete zakazati konsultacije sa genetskim savetovalištem Premium Genetics-a, a za više informacije pozovite 064 659 24 28.
Zašto je važno uraditi SMA PANEL?
- genetski test koji budućim roditeljima omogućava da utvrde da li su nosioci gena za SMA i postoji li rizik za prenošenje gena ili oboljenja na potomstvo
- SMA je retka, ali vrlo teška i progresivna bolest mišića i vodeći je genetski uzrok smrtnosti dece u najranijem uzrastu
- Smatra se da je jedna od 50 osoba nosilac gena za SMA. Nosioci mutiranih gena nemaju oboljenje, ni ne znaju da poseduju mutirani gen, ali mogu svom potomstvu preneti mutaciju koja dovodi do SMA
- Pokrivene su sve regije od interesa za SMA
- U zemljama gde testiranje na SMA još uvek nije deo nacionalnog programa neonatalnog skrininga, kao što je slučaj sa našom zemljom, roditeljima su dostupni testovi za tzv. skrining nosilaca bolesti, koji se mogu uraditi pre začeća, tokom trudnoće ili po rođenju deteta.
Uz svaki panel moguće je dodatno uraditi analizu na SMA
Najčešće
POSTAVLJANA PITANJA?
Neinvazivni prenatalni test (NIPT) spada u grupu tzv. skrining testova koji analizira slobodnocirkulišuću DNK fetusa, sa ciljem otkrivanja raznih genetskih abnormalnosti. Saznajte još detaljnije o tome šta je neinvazivni prenatalni test.
Hromozomi su strukture karakterističnog oblika koja se dobro boje (naziv potiče od gr. chromos = boja i soma = telo). U jedru se mogu uočiti za vreme deobe. Hromozome je prvi otkrio naučnik Valter Fleming (1843-1905) 1882. godine, prilikom istraživanja deobe ćelija. Najbolje se uočavaju za vreme metafaze mitoze, pa se tada i izučavaju i nazivaju metafazni hromozomi ili mitotski hromozomi.
Aneuploidije polnih hromozoma su genetske bolesti uzrokovane prisustvom ili odsustvom polnog hromozoma. Inače, 23. par hromozoma određuje pol individue. Žene imaju dva X hromozoma, a muškarci jedan X i jedan Y hromozom.
Postoje četiri velike aneuploidije polnih hromozoma:
- Tarnerov sindrom se karakteriše prisustvom jednog X hromozoma;
- Trostruki X sindrom se karakteriše prisustvom tri X hromozoma;
- Klinefelterov sindrom se karakteriše prisustvom dva X i jednog Y hromozoma;
- Jakobsov sindrom se karakteriše prisustvom jednog X i dva Y hromozoma.
Razgovarajte sa Vašim doktorom o primeni Premium Genetics prenatalnog testa u cilju detekcije fetalnih hromozomskih aneuploidija.
U genetici je preciznost veoma važna, tako da se poslovica “od viška glava ne boli” ne može primeniti. Naime, ukoliko u ćeliji postoje tri hromozoma (trizomik) umesto jednog para (dizomik), takođe dolazi do aneuploidije – u ovom slučaju jer je broj hromozoma 47 umesto 46. Trizomija može pogoditi bilo koji hromozomski par, ali nije uvek fatalna, kao što je to slučaj sa monozomijom. U zavisnosti od mesta na kojem se nalaze, trizomije mogu dovesti do problema u rastu i razvoju ploda, do pojave zdravstvenih problema i karakterističnih fizičkih obeležja, a mogu uticati i na dužinu i kvalitet života. Daunov, Edvardsov i Patau sindrom su trizomije koje se najčešće sreću kod čoveka i kompatibilne su sa životom postpartum. Saznajte više.
Daunov sindrom je najčešća trizomija u opštoj populaciji. Učestalost Daunovog sindroma je 1:700 kod novorođene dece. Na učestalost Daunovog sindroma utiču pre svega godine majke, tako da se rizik za postojanje Daunovog sindroma povećava kod trudnica koje imaju preko 35 godina. Međutim, to ne znači da su mlađe trudnice u kategoriji koja je bez rizika. U poslednje vreme, sve je veći broj mlađih trudnica, kod kojih se u trudnoći detektuje plod sa Daunovim sindromom. Saznajte više.
Pored Daunovog i Edvardsovog sindroma, Patau sindrom je jedna od najučestalijih trizomija koje se javljaju kod čoveka. Do greške u kopiranju genetičkog materijala dolazi u ranim fazama trudnoće, a rezultat nepravilne ćelijske deobe je, u ovom slučaju, nastanak dodatne kopije celog ili dela hromozoma 13. Posledica je da plod umesto tipične 2 ima 3 kopije ovog hromozoma. Dakle, ćelije ploda umesto tipičnih 46, broje 47 hromozoma. U zavisnosti od mesta na kojem se nalaze, trizomije mogu dovesti do problema u rastu i razvoju ploda, do pojave zdravstvenih problema i karakterističnih fizičkih obeležja, a mogu uticati i na dužinu i kvalitet života. Trizomija 13 utiče na rast i razvoj bebe in utero (u materici), a izaziva i brojne zdravstvene i razvojne probleme po rođenju.
Sindrom nosi ime po nemačkom naučniku Klausu Patau, koji je 1960. godine, zajedno sa svojim saradnicima, opisao simptome i hromozomsku prirodu sindroma. Statistički podaci pokazuju da je incidenca 1:10.000 novorođenih beba. Saznajte više.
Tipična ljudska jedinka ima 46 hromozoma, koji su organizovani u 23 para, a nasleđuje ih od svojih roditelja. Da bi od dva biološki zdravlja roditelja nastalo biološki zdravo dete, kopiranje genetičkog materijala se mora odviti bez greške. Bilo kakve greške u kopiranju, mogu rezultirati ozbiljnim odstupanjima u kreiranju genotipa nove jedinke, što za posledicu može imati pojavu raznih zdravstvenih problema ili fizičkih i intelektualnih abnormalnosti.
Dakle, da bi se plod od samog začeća normalno razvijao, uslov je precizno kopiranje genetičkog materijala roditelja. Čovek ima 23 para hromozoma, različtih veličina i oblika, a svi su pažljivo numerisani međunarodnom konvencijom. Prva 22 para nazivamo autozomnim, dok poslednji par određuje pol deteta. Bilo kakva odstupanja u kopiranju genetičkog materijala u parovima hromozoma, rezultiraće različitim oboljenjima ili poremećajima. Do greške u kopiranju genetičkog materijala dolazi u veoma ranoj trudnoći – nekad već prilikom spajanja spermatozoida i jajne ćelije, a nekad u ranoj fazi razvoja embriona. Dodatna kopija genetičkog materijala (3 umesto 2 kopije) dovešće do trizomija, dok će nedostatak genetičkog materijala (1 umesto 2 kopije) dovesti do monozomija. Edvardsov sindrom ili trizomija 18, zajedno sa Daunovim i Patau sindromom, predstavlja jednu od najučestalijih trizomija kod čoveka.
Edvardsov sindrom ili trizomija 18 nastaje (kako i samo ime kaže) kada se dodatna kopija genetičkog materijala pojavi na 18. hromozomu. Ime nosi po engleskom genetičaru Edvardsu (John Hilton Edwards), koji je prvi opisao sindrom 1960. godine. Saznajte više.
Premium Genetics test je neinvazivni prenatalni test najnovije generacije, koji precizno detektuje Daunov, Edvardsov i Patauov sindrom, tri najčešća fetalna genetska poremećaja, kao i druge sindrome uzrokovane aneuploidijama X i Y hromozoma.
Neinvazivno prenatalno testiranje je važno, jer omogućava bezbedan i precizan način testiranja mogućih genetskih poremećaja, od kojih je najčešći Daunov sinrom, kod bebe i to pre rođenja. Pre uvođenja ovog revolucionarnog testa, opcije za testiranje Daunovog sindroma su bile ili skrining metode poput ultrazvuka kombinovanog sa ostalim laboratorijskim testovima koja je imala nisku preciznost (približno 80-95%) ili prenatalna dijagnostika visoke preciznosti koja koristi invazivne metode (npr. amniocinteza ili uzorkovanje horionskih čupica (CVS)) sa 0.5% rizika od pobačaja. Premium Genetics test je neinvazivni prenatalni test najnovije generacije, koji ima značajno veću preciznost (>99%) bez rizika od pobačaja. Saznajte više.
Šta su MONOGENSKE BOLESTI?
Monogenske bolesti su oboljenja koja nastaju usled prisustva mutacije (promjene) na jednom genu. Gen na kome se nalazi ta promena (mutirani gen) može biti smešten na autozomnim hromozomima (hromozomi 1-22) ili polnim hromozomima (X i Y hromozomi). Svaka osoba poseduje mutacije u svojim genima. Međutim, nisu sve mutacije patološke, tj. neće svaka mutacija gena dovesti do razvoja nekog oboljenja. Autozomi, tj. telesni hromozomi nose gene koji određuju telesne osobine. Polni hromozomi su hromozomi na kojima se nalaze geni koji određuju pol, kao i funkciju pola.
Različiti oblici jednog istog gena nazivaju se aleli. Potomci nasleđuju po jedan alel od svakog roditelja – jedan od oca i jedan od majke. Kombinovanjem dve različite garniture gena (jedna od oca i jedna od majke) nastaju određene osobine potomaka – boja očiju, boja kose, visina, oblik nosa i druga fizička obeležja. Na isti ovaj način, roditelji svojoj deci mogu preneti i određena oboljenja.
Roditelji često mogu biti nosioci samo jednog mutiranog genskog alela i u tom slučaju se bolest ne ispoljava kod roditelja. S obzirom na to da tada kod roditelja ne postoje nikakvi klinički znakovi i simptomi bolesti, u najvećem broju slučajeva, roditelji ne mogu ni znati da su nosioci mutacije na nekom od gena. Roditelji koji su nosioci mutiranog genskog alela mogu taj promenjeni (mutirani) alel prenositi dalje na svoje potomke i na taj način se mutacija može prenositi kroz generacije.

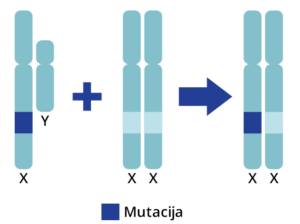
Monogenske bolesti koje su obuhvaćene PremiumGene testom nasleđuju se autozomno recesivno ili recesivno vezano za X hromozom.
Kod bolesti koje se nasleđuju autozomno recisivno, ukoliko su oba roditelja nosioci iste mutacije na određenom genu, postoji verovatnoća od 25% da dete bude potpuno zdravo, 25% je verovatnoća da dete ima oboljenje i 50% verovatnoće da dete bude zdravo, ali da je nosilac iste mutacije koju može prenositi na svoje potomstvo.
Kod recesivnog nasleđivanja vezanog za X-hromozom, žene su prenosioci bolesti, dok uvek isključivo oboljevaju muškarci, odnosno muška deca. Za razliku od autozomno recesivnog nasleđivanja, oboleli muškarac gotovo nikad ne prenosi mutaciju svojim muškim potomcima. Međutim, svi ženski potomci obolelog muškarca su prenosioci mutacije.
Targetirana genomska analiza
Premium Genetics prenatalni testovi koriste sopstvene patentne, ciljane sekvence dizajnirane da izbegnu sekvence sa varijabilnim brojem kopija (CNVs), ponavljajuće elemente DNK i kompleksnu arhitekturu genoma. Ovaj ciljani pristup prevazilazi probleme povezane sa drugim NIPT-ovima i povećava preciznost i tačnost ovog testa. To donosi najmanje lažno pozitivnih rezultata.
Merenje fetalne frakcije
Premium Genetics testovi zahvaljujući savremenoj tehnologiji pouzdano razdvajaju cfDNK bebe od cfDNK majke. Patentni bioinformatički softver koristi tačke visoke dubine čitanja ovih informativnih lokusa za precizno računanje fetalne frakcije. Precizne mjere fetalne frakcije povećavaju preciznost i pouzdanost prenatalnog testa.
Šta testira
PremiumGene test?

Pol bebe
Koje monogenske bolesti analizira
PremiumGene test?
3-HMGCL deficit nastaje usled mutacije HMGCL gena i nasleđuje se autozomno recesivno, što znači da roditelji mogu biti nosioci samo jednog mutiranog genskog alela, ali u tom slučaju ne pokazuju kliničke znakove i simptome bolesti (čak i ne znaju da su nosioci mutacije). Međutim, ukoliko potomci naslede oba mutirana genska alela od svojih roditelja može doći do ispoljavanja bolesti.
Osnovna karakteristika je metabolička acidoza zbog promene pH krvi usled nakupljanja štetnih produkata. Nizak nivo šećera u krvi direkto pogađa ćelije mozga. Simptomi bolesti obično nastaju već u prvoj godini života. 3-HMGCL deficit uzrokuje povraćanje, dijareju, dehidrataciju, umor, oslabljen mišićni tonus. Ukoliko se ne leči izaziva poremećaj disanja, konvulzije, komu pa čak i smrtni ishod.
Nasledni poremećaj u metabolizmu proteina, usled deficita proteina uzrokovanog mutacijom MCCC1 gena. Nasleđuje se autozomno recesivno, što znači da roditelji mogu biti nosioci samo jednog mutiranog genskog alela, ali u tom slučaju ne pokazuju kliničke znakove i simptome bolesti (čak i ne znaju da su nosioci mutacije). Međutim, ukoliko potomci naslede oba mutirana genska alela od svojih roditelja može doći do ispoljavanja bolesti.
Javlja se poremećaj u metabolizmu proteina zbog nemogućnosti obrade leucina. Toksični bioprodukti se akumuliraju u mozgu. Učestalost javljanja je 1 na 36000 novorođenih. Klinička slika varira od blage do vema teške, koja ugrožava život. Otežano hranjenje, povraćanje, dijareja, hipotonija, letargija su najčešće kliničke manifestacije.
Nasledni poremećaj u metabolizmu proteina, usled deficita proteina uzrokovanog mutacijom MCCC2 gena. Nasleđuje se autozomno recesivno, što znači da roditelji mogu biti nosioci samo jednog mutiranog genskog alela, ali u tom slučaju ne pokazuju kliničke znakove i simptome bolesti (čak i ne znaju da su nosioci mutacije). Međutim, ukoliko potomci naslede oba mutirana genska alela od svojih roditelja može doći do ispoljavanja bolesti.
Javlja se poremećaj u metabolizmu proteina zbog nemogućnosti obrade leucina. Toksični bioprodukti se akumuliraju u mozgu. Učestalost javljanja je 1 na 36000 novorođenih. Klinička slika varira od blage do vema teške, koja ugrožava život. Otežano hranjenje, povraćanje, dijareja, hipotonija, letargija su najčešće kliničke manifestacije.
Nasleđuje se autozomno recesivno, što znači da roditelji mogu biti nosioci samo jednog mutiranog genskog alela, ali u tom slučaju ne pokazuju kliničke znakove i simptome bolesti (čak i ne znaju da su nosioci mutacije). Međutim, ukoliko potomci naslede oba mutirana genska alela od svojih roditelja može doći do ispoljavanja bolesti.
Usled mutacije MTTP (mikrozoomalni protein za transport triglicerida) nastaje deficijencija apolipoproteina B48 i B100 koji su potrebni za stvaranje hilomikrona VLDL. Nastaje poremećaj apsorcije masti i vitamina rastvorljivih u masti iz hrane. Klinička slika obuhvata nenapredovanje, steatoreju, penušave stolice, distenziju stomaka, intelektualni deficit, slabost mišića, skoliozu, progresivan gubitak vida. Nelečena bolest dovodi do ozbiljnih neuroloških deficita, može dovesti do smrti.
Deficit akril-CoA oksidaze I nastaje usled mutacije ACOX1 gena i nasleđuje se autozomno recesivno, što znači da roditelji mogu biti nosioci samo jednog mutiranog genskog alela, ali u tom slučaju ne pokazuju kliničke znakove i simptome bolesti (čak i ne znaju da su nosioci mutacije). Međutim, ukoliko potomci naslede oba mutirana genska alela od svojih roditelja može doći do ispoljavanja bolesti.
Poremećaj se ispoljava već na samom rođenju, prvenstveno je zahvaćen nervni sistem. Većina novorođenčadi ima oslabljen mišićni tonus, neobične crte lica, nisko postavljene ušne školjke. Pojava šestog prsta ili uvećena jetra takođe se javlja kod nekih osoba. Psihomotorni razvoj je odložen što znači da deca kasnije počinju da govore i hodaju, ali postepeno doživljavaju gubitak naučenih veština (razvojna regresija). Kako se stanje pogoršava povećan je tonus mišića (hipertonija), ponavljaju se napadi epilepsije uz pogoršanje vida i sluha.
Aicardi-Goutières Syndrome može imati različite oblike nasleđivanja. Mutacija na SAMDH1 genu nasleđuje se autozomno recesivno, što znači da roditelji mogu biti nosioci samo jednog mutiranog genskog alela, ali u tom slučaju ne pokazuju kliničke znakove i simptome bolesti (čak i ne znaju da su nosioci mutacije). Međutim, ukoliko potomci naslede oba mutirana genska alela od svojih roditelja može doći do ispoljavanja bolesti.
SAMDH1 gen kodira sintezu proteina čije su komponente bitne za replikaciju i održavanje genetskog materijla (DNK) u ćelijama. Istraživanja pokazuju da mutacije (promene) u SAMHD1 genu mogu dovesti do proizvodnje SAMDH1 proteina čije komponente oštećuju DNK u ćelijama. Kao rezultat toga aktivira se imuni sistem koji napada sopstvena tkiva izazivajući upalno oštećenje mozga, kože i dugih telesnih sistema.
Aicardi-Goutières Syndrome (AGS Sindrom) je sindrom koji prvenstveno pogađa mozak. Većina novorođenčadi ne pokazuje znakove ili simptome poremećaja na rođenju. Međutim, oko 20% može imati uvećenu jetru, slezinu, povišen nivo jetrenih enzima , smanjen broj trombocita i neurološke abnormalnosti. Već u prvoj godini života doživljavaju epizode teške disfunkcije mozga (encefalopatija). Postepeno prestaju razvijati nove veštine ili gube već stečene što je posledica trajnog neurološkog oštećenja koje je veoma ozbiljno. Usporen je rast lobanje i mozga što rezultira malom veličinom glave (mikrocefalija). Pojedine osobe sa ovim sindromom imaju i karakteristike koje su specifične za autoimune poremećaje.
Pojedini oblici bolesti mogu biti blaži i nastati u kasnijem životnom dobu, dok se teži oblici bolesti završavaju letalno već u periodu detinjstva.
Alportov sindrom može imati različite oblike nasleđivanja, a najčešće je (u 85% slučajeva) vezano za X hromozom. Pogađa 1 na 5.000-10.000 dece.
Mutacijom gena COL4A5 nastaje teški progresivni oblik nefritisa i senzorineuralno oštećenje sluha. Klinički se ispoljava već u ranom detinjstvu pojavom mikrohematurije (prisustvo krvi u mokraći) ili proteinurije (prisustvo proteina u mokraći). Kod polovine obolelih nakon 10. godine života javlja se nefrotski sindrom praćen hipertenzijom, dok postepeno dolazi do razvoja bubrežne insuficijencije. Lečenje bolesti je simptomatsko, dok u stadijumu terminalne hronične bubrežne insuficijencije (završni stadijum bolesti) primenjuje se dijaliza i transplantacija bubrega.
Gubitak sluha je obostran, najčešće se otkriva do 10. godine koja može biti progresivna. Teži stepen gluvoće zahteva primenu slušnog aparata.
Mutacija ALMS1 gena koja je odgovorna za nastanak Alstromovog sindroma nasleđuje se autozomno recesivno, što znači da roditelji mogu biti nosioci samo jednog mutiranog genskog alela, ali u tom slučaju ne pokazuju kliničke znakove i simptome bolesti (čak i ne znaju da su nosioci mutacije). Međutim, ukoliko potomci naslede oba mutirana genska alela od svojih roditelja može doći do ispoljavanja bolesti.
Alstromov sindrom počinje još u ranom detinjstvu, mada se neki oblici mogu pojaviti i kasnije u životu. Klinički se ispoljava progresivnim gubitkom vida i sluha, niskim rastom, dilatativnom kardiomiopatijom, gojaznošću, a češće se javlja dijabetes melitus tip 2. Mogu biti zahvaćeni i drugi organi kao što su jetra, bubrezi, bešika, pluća što može imati fatalne posledice.
Andermanov sindrom je poremećaj koji nastaje usled mutacije gena SLC12A6 i nasleđivanje je autozomno recesivno, što znači da roditelji mogu biti nosioci samo jednog mutiranog genskog alela, ali u tom slučaju ne pokazuju kliničke znakove i simptome bolesti (čak i ne znaju da su nosioci mutacije). Međutim, ukoliko potomci naslede oba mutirana genska alela od svojih roditelja može doći do ispoljavanja bolesti.
SLC12A6 gen odgovoran je za transport jona kalijuma (K) i hlora (Cl) kroz ćelijsku membranu. Pozitivno naelektrisani joni kalijuma i negativno naelektrisani joni hlora prenose se istovremeno tako da omogućavaju nastanak elektroneutralne sredine što je bitno za održavanje mnogih telesnih funkcija. Smatra se da nedostatak funkcionalnog proteina ometa razvoj corpusa callosuma i nerava kojma se prenose nervni impulsi za kretanje i osećaj što rezultira znacima i simptomima Andermanovog sindroma.
Osobe pogođene sa ovim sindromom imaju abnormalne ili odsutne reflekse i slab mišićni tonus, takođe ozbiljnu progresivnu slabost i gubitak osećaja u ekstremitetima. Obično prohodaju između 3. i 4. godine, ali tu sposobnost gube u tinejdžerskim godinama. U kasnijem životnom dobu razvijaju deformacije zglobova koje ograničavaju kretanje. Često su zahvaćeni i karanijalni nervi što dovodi do slabosti mišića lica. Ostali simptomi su: psihički poremećaji, hipertelorizam, široka i kratka lobanja. Sindrom je povezan sa kraćim životnim vekom.
Deficit aromataze koji nastaje zbog mutacije CYP19A1 gena nasleđuje se autozomno recesivno, što znači da roditelji mogu biti nosioci samo jednog mutiranog genskog alela, ali u tom slučaju ne pokazuju kliničke znakove i simptome bolesti (čak i ne znaju da su nosioci mutacije). Međutim, ukoliko potomci naslede oba mutirana genska alela od svojih roditelja može doći do ispoljavanja bolesti.
Enzim aromataza je presudan u biosintezi estrogena iz androgena. Manjak funkcionalne aromataze dovodi do smanjene proizvodnje estrogena i povećanja nivoa androgena.
Žene sa nedestatkom aromataze imaju netipičan izgled spoljašnjih genitalija, ali normalan izgled unutrašnjih reproduktivnih organa. U ranom detinjstvu javljaju se ciste jajnika, dok u periodu adolesencije većina ne razvija sekundarne seksualne karakteristike poput rasta grudi. Sklone su razvoju akni i prekomernom rastu dlačica na telu (hirzutizam).
Muškarci sa ovim stanjem imaju očuvan izgled spoljašnjih muških genitalija, dok neke prati smanjen seksualni nagon, poremećaj proizvodnje i sastava semene tečnosti, mali i nespušteni testis.
Druge karakteristike povezane sa nedostatkom aromataze koje se javljaju kod oba pola su: visok rast, osteoporoza, povišen nivo šećera u krvi, gojaznost.
Autozomno recesivno oboljenjekoje je izazvano mutacijom gena SLC35a3, što znači da roditelji mogu biti nosioci samo jednog mutiranog genskog alela, ali u tom slučaju ne pokazuju kliničke znakove i simptome bolesti (čak i ne znaju da su nosioci mutacije). Međutim, ukoliko potomci naslede oba mutirana genska alela od svojih roditelja može doći do ispoljavanja bolesti.
Kliničke manifestacije: mikrocefalija, artrogripoza, kontrakture zglobova, dislokacije kukova, displazija acetabuluma, dislokacija kolena, fleksione kontrakture prstiju šake i stopala, devijacije i deformacije istih, hipotonija, usporen psihomotorni razvoj, mentalna retardacija, konvulzije, autizam.
Deficit asparagin sintetaze koji nastaje zbog mutacije ASNS gena nasleđuje se autozomno recesivno, što znači da roditelji mogu biti nosioci samo jednog mutiranog genskog alela, ali u tom slučaju ne pokazuju kliničke znakove i simptome bolesti (čak i ne znaju da su nosioci mutacije). Međutim, ukoliko potomci naslede oba mutirana genska alela od svojih roditelja može doći do ispoljavanja bolesti.
Asparagin sintetaza je enzim koji se nalazi u ćelijama čitavog tela i odgovoran je za nastanak aminokiseline asparagin koja je važan neurotransmiter za prenos signala u mozgu.
Neurološki problem počinju ubrzo nakon rođenja, dok većina ima neobično malu veličinu glave (mikrocefalija), vremenom dolazi i do atrofije mozga. Posledica je ozbiljno kašnjenje u razvoju koje utiče na mentalne i motoričke aktivnosti. Mogu biti pojačani mišićni refleksi i slab tonus mišića, česti su epileptični napadi. Usled atrofije mozga može nastati slepilo (kortikalno slepilo). Sindrom je povezan sa kraćim životnim vekom.
Aspartilglukozamiunurija nastaje usled mutacije AGA gena i nasleđuje se autozomno recesivno, što znači da roditelji mogu biti nosioci samo jednog mutiranog genskog alela, ali u tom slučaju ne pokazuju kliničke znakove i simptome bolesti (čak i ne znaju da su nosioci mutacije). Međutim, ukoliko potomci naslede oba mutirana genska alela od svojih roditelja može doći do ispoljavanja bolesti.
AGA gen kodira sintezu enzima aspartilglukozaminidaze u lizozomima. Lizozomi su ćelijske organele poput reciklažnih centara u kojima enzim razlaže molekule glikoproteina. Mutacija AGA gena dovodi do neadekvatne aktivnosti navedenog enzima.
Nakupljanje glikoproteina posebno utiče na nervne ćelije u mozgu što je uzrok njihovog propadanja i pada mentalnog funkcionisanja.
Genetski autozomno recesivni poremećaj usled mutacije PKHD1 gena, što znači da roditelji mogu biti nosioci samo jednog mutiranog genskog alela, ali u tom slučaju ne pokazuju kliničke znakove i simptome bolesti (čak i ne znaju da su nosioci mutacije). Međutim, ukoliko potomci naslede oba mutirana genska alela od svojih roditelja može doći do ispoljavanja bolesti.
Karakteriše ga stvaranje cista u bubregu i jetri, problemi u drugim organima poput mozga i srca. Ispoljavanje je varijabilno, pa može ostati neispoljena i posle detinjstva. Neka deca umiru u prvim satima ili danima po rođenju, usled respiratorne insuficijencije ili bubrežna insuficijencija nastaje kasnije tokom života. Kauzalna terapija još nije pronađena.
Bardet Biedl sindrom može nastati kao rezultat mutacija u najmanje 14 različitih gena. Skoro 1/4 svih slučajeva nastaje kao rezultat mutacije BBS1 gena i nasleđuje se autozomno recesivno, što znači da roditelji mogu biti nosioci samo jednog mutiranog genskog alela, ali u tom slučaju ne pokazuju kliničke znakove i simptome bolesti (čak i ne znaju da su nosioci mutacije). Međutim, ukoliko potomci naslede oba mutirana genska alela od svojih roditelja može doći do ispoljavanja bolesti.
Mutacije BBS gena dovode do poremećaja strukture i pravilnog funkcionisanja cilija što narušava važne puteve hemijske signalizacije i poremećaja percepcije. Cilije su izuzetno važne strukture za percepciju čula (poput vida, sluha, mirisa).
Gubitak vida je glavno obeležje Bardet Biedlovog sindroma. Najpre se tokom detinjstva u vidnom polju javljju slepe mrlje koje se postepeno spajaju i proširuju, te stvaraju tunelski vid do perioda adolescencije. Ostali simptomi i znakovi bolesti su: prisustvo dodatnih prstiju (polidaktilija), poremećaj intelektualnog razvoja i problemi sa učenjem, smanjen nivo polnih hormona, poremećaji bubrega koji su mogu biti opasni po život.
Nasledno oboljenje koje zahvata mnoge delove organizma. Nastaje kao rezultat mutacije BBS12 gena i nasleđuje se autozomno recesivno, što znači da roditelji mogu biti nosioci samo jednog mutiranog genskog alela, ali u tom slučaju ne pokazuju kliničke znakove i simptome bolesti (čak i ne znaju da su nosioci mutacije). Međutim, ukoliko potomci naslede oba mutirana genska alela od svojih roditelja može doći do ispoljavanja bolesti.
Osobe sa ovim sindromom imaju progresivan poremećaj vida, polidaktiliju, centralnu gojaznost, hipogonadizam, bubrežne abnormalnosti, teškoće u učenju, moguća je pojava dijabetesa, hipertenzije, srčanih defekata, bolesti creva, poremećaj govora, poremećaj ponašanja, abnormalnosti zuba. Terapija nije pronađena.
Nasledni poremećaj usled mutacije HBB gena i nasleđuje se autozomno recesivno, što znači da roditelji mogu biti nosioci samo jednog mutiranog genskog alela, ali u tom slučaju ne pokazuju kliničke znakove i simptome bolesti (čak i ne znaju da su nosioci mutacije). Međutim, ukoliko potomci naslede oba mutirana genska alela od svojih roditelja može doći do ispoljavanja bolesti.
Zbog mutacije gena poremećena je struktura hemoglobina. Težina kliničke slike varira usled stepena nedostatka beta jedinice globina. Ovo je uzrok neadekvatne funkcije hemoglobina i kraćeg života eritrocita što dovodi do anemije, koja uzrokuje slabost, umor, bledilo kože, embolizacije krvnh sudova, nenapredovanja deteta, deformacije koštanog sistema žutice, uvećanja jetre, slezine, srca. Ova deca mogu zahtevati učestale transfuzije krvi, što u nekim slučajevima dovodi do nagomilavanja gvožđa u organizmu sa svojim posledicama.
Deficit biotinidaze nastaje usled mutacije BTD gena i nasleđuje se autozomno recesivno, što znači da roditelji mogu biti nosioci samo jednog mutiranog genskog alela, ali u tom slučaju ne pokazuju kliničke znakove i simptome bolesti (čak i ne znaju da su nosioci mutacije). Međutim, ukoliko potomci naslede oba mutirana genska alela od svojih roditelja može doći do ispoljavanja bolesti.
U normalnim okolnostima, enzim biotinidaza oslobađa vitamin biotin (B7) iz njegovog vezanog oblika i time ga organizam koristi za proizvodnju drugih enzima, što rezultira smetnjama u procesu glukoneogeneze i lipogeneze.
Simptomi nedostatka biotina su: gubitak kose,, konjuktivitis, depresija, letargija, halucinacije, osećaj trnjenja i pečenja u ekstremitetima, bledilo, mučnina, povraćanje, dermatitis.
Kanavanova bolest nastaje usled mutacije ASPA gena i nasleđuje se autozomno recesivno, što znači da roditelji mogu biti nosioci samo jednog mutiranog genskog alela, ali u tom slučaju ne pokazuju kliničke znakove i simptome bolesti (čak i ne znaju da su nosioci mutacije). Međutim, ukoliko potomci naslede oba mutirana genska alela od svojih roditelja može doći do ispoljavanja bolesti.
Deficijencija enzima Aspart acilaze dovodi do akumulacije N-Acetil aspartične kiseline u mozgu i oštećenja bele mase mozga sa stvaranjem sunđerastog tikva (leukodistrofija). Klinički se ispoljava u prvoj godini života. Primećuje se poremećaj tonusa svih mišića (dete ne počinje da diže glavu), naglo nastaje makrocefalija, nastaju problemi sa uzimanjem hrane, vraćanje hrane na nos, gubitak vida i sluha, epileptični napadi. Ova deca ne mogu da puze, sede, hodaju i pričaju. Vremenom nastaju paralize, mentalna retardacija i slepilo. Većina dece ne živi duže od 10 godina. Kauzalna terapija ne postoji.
Karpenterov sindrom nastaje zbog mutacije RAB23 gena i nasleđuje se autozomno recesivno, što znači da roditelji mogu biti nosioci samo jednog mutiranog genskog alela, ali u tom slučaju ne pokazuju kliničke znakove i simptome bolesti (čak i ne znaju da su nosioci mutacije). Međutim, ukoliko potomci naslede oba mutirana genska alela od svojih roditelja može doći do ispoljavanja bolesti.
RAB23 gen kodira informacije za prenos proteina i ostalih molekula unutar ćelija što je od izuzetne važnosti za rast ćelija (proliferaciju), njihovu diferencijaciju i oblikovanje mnogih delova tela.
Karpenterov sindrom karakteriše prerana fuzija određenih kostiju lobanje (kraniosinostoza), takođe i fuzija kože između 2 ili više nožnih prstiju, neobično kratki ili dodatni prsti nogu. Kraniosinostoza sprečava normalan rast lobanje što joj daje naglašen izgled i često izraženu asimetriju lica. Rana fuzija kostiju lobanje može uticati na razvoj mozga i dovodi do povećanja pritiska unutar lobanje. Često imaju intelektualni poremećaj različitog stepena, dok pojedinci imaju očuvanu intelektualnu sposobnost. Organi u grudima i trbuhu mogu imati nekarakterističan položaj (npr. srce je postavljeno na desnu stranu, umesto na levu).
Nasledno oboljenje koje nastaje usled mutacije gena VDS13a, koji je odgovoran za stvaranje proteina horeina. Nasleđuje se autozomno recesivno, što znači da roditelji mogu biti nosioci samo jednog mutiranog genskog alela, ali u tom slučaju ne pokazuju kliničke znakove i simptome bolesti (čak i ne znaju da su nosioci mutacije). Međutim, ukoliko potomci naslede oba mutirana genska alela od svojih roditelja može doći do ispoljavanja bolesti.
Ispoljava se u detinjstvu ili ranom odraslom dobu poremećajima pokreta (horea), poremećajem nerava i mišića (distonija), otežanim govorom i hranjenjem, spazmom mišića. Postoji abnormalnost eritrocita – akantociti. Otežano učenje, pamćenje, ponašanje. Kod polovine dolazi do pojave epileptičnih napada. Vremenom se pogoršava.
Horoideremija je oboljenje koje nastaje usled mutacije CHM gena i naleđuje se recesivno vezano za X hromozomom.
Glavna odlika je progresivni gubitak vida. Kao prvi simptom ovog stanja obično se javlja oštećenje noćnog vida (noćno slepilo), koje se može javiti već u ranom detinjstvu. Sledi progresivno sužavanje vidnog polja (tunelski vid) i smanjena oštrina vida. Problemi sa vidom nastaju usled gubitka ćelija u specijalizovanom tkivu osetljivom na svetlost. Međutim, svi pojedinci će u kasnijem životnom dobu razviti slepilo.
Smatra se da je horoideremija u oko 4% uzrok slepila.
Citrulinemija tip II nastaje usled mutacije SLC25A13 gena, odgovornog za proizvodnju citrina i nasleđuje se autozomno recesivno, što znači da roditelji mogu biti nosioci samo jednog mutiranog genskog alela, ali u tom slučaju ne pokazuju kliničke znakove i simptome bolesti (čak i ne znaju da su nosioci mutacije). Međutim, ukoliko potomci naslede oba mutirana genska alela od svojih roditelja može doći do ispoljavanja bolesti.
Citrin je protein koji je aktivan u jetri, bubrezima i srcu, odgovoran za transport molekula kroz mitohondrije. Ovaj protein igra ulogu u proizvodnji i razgradnji prostih šećera, a takođe i u proizvodnji nukleotida koji su sastavni deo DNK molekula. Mutacija SLC25A13 gena dovodi do proizvodnje nestabilnog citrina koji se brzo razgrađuje.
Citrulinemiju tip II izaziva neurološke probleme kod odraslih kao što su zbunjenost, nemir, razdražljivost, epizode napada. Mutacije SLC25A13 gena pronađene su i kod novorođenčadi sa poremećajem jetre (neonatalna intrahepatička holestaza izazvana nedostatkom citrina) gde je poremećen protok žuči, što smanjuje apsorpciju hranljivih materija.
Kombinovani nedostatak enzima oksidativne fofsorilacije nastaje zbog mutacije TSFM gena i nasleđuje se autozomno recesivno, što znači da roditelji mogu biti nosioci samo jednog mutiranog genskog alela, ali u tom slučaju ne pokazuju kliničke znakove i simptome bolesti (čak i ne znaju da su nosioci mutacije). Međutim, ukoliko potomci naslede oba mutirana genska alela od svojih roditelja može doći do ispoljavanja bolesti.
Radi se poremećaju koji je praćen teškom metaboličkom acidozom sa encefalomiopatijom ili hipertrofičnom kardiomiopatijom.
Najčešći tipovi urođenih poremećaja glikozilacije su IA I IC.
Tip IA nastaje kao posledica smanjene aktivnosti enzima fosfomanomutaze 2, usled mutacije PMM2 gena. Nasleđivanje je autozomno recesivno, što znači da roditelji mogu biti nosioci samo jednog mutiranog genskog alela, ali u tom slučaju ne pokazuju kliničke znakove i simptome bolesti (čak i ne znaju da su nosioci mutacije). Međutim, ukoliko potomci naslede oba mutirana genska alela od svojih roditelja može doći do ispoljavanja bolesti.
Zahvata više organskih sistema i klinička slika je vrlo heterogena Najčešći simptomi su: mentalna retardacija, teško zaostajanje u psihomotornom razvoju, konvulzije, poremećaj nervog sistema, fibroza jetre, poremaćaji koagulacije.
Tešku kongenitalnu neutropeniju prvi put je opisao 1956. godine švedski pedijatar Rolf Kostmann. Sada je poznato da se radi o heterogenom poremećaju koji se u 30% slučajeva nasleđuje autozomno recesivno usled mutacije HAX1 gena. To znači da roditelji mogu biti nosioci samo jednog mutiranog genskog alela, ali u tom slučaju ne pokazuju kliničke znakove i simptome bolesti (čak i ne znaju da su nosioci mutacije). Međutim, ukoliko potomci naslede oba mutirana genska alela od svojih roditelja može doći do ispoljavanja bolesti.
Novorođenčad prati izrazito smanjen broj neutrofila već na samom rođenju, što dovodi to teških bakterijskih infekcija. U prvih nekoliko meseci života javljaju se visoke temperature, infekcije kože, gingivitisi, stomatitisi, upale pluća i često su ozbiljne infekcije bile uzrok smrtnosti u periodu do 1. godine. Ozbiljno stanje koje zahteva dodatno praćenje zbog opasnosti od razvoja mijelodisplazije i akutne mijeloidne leukemije.
Krigler Najarov (Crigler-Najjar) sindrom tip 1 je nasledno autozomno recesivno oboljenje, što znači da roditelji mogu biti nosioci samo jednog mutiranog genskog alela, ali u tom slučaju ne pokazuju kliničke znakove i simptome bolesti (čak i ne znaju da su nosioci mutacije). Međutim, ukoliko potomci naslede oba mutirana genska alela od svojih roditelja može doći do ispoljavanja bolesti.
Ispoljava se visokim koncentracijama nekonjugovanog bilirubina u krvi. To je zapravo poremećaj metabolizma bilirubina, koji se hemijski formira razlaganjem hema u crvenim krvnim zrncima. Poremećaj dovodi do formiranja nehemolitičke žutice, pri kojoj se javljaju visoki nivoi nekonjugovanog bilirubina i često dolazi do oštećenja mozga kod odojčadi.
Nasleđuje se autozomno recesivno usled mutacije CFTR gena, što znači da roditelji mogu biti nosioci samo jednog mutiranog genskog alela, ali u tom slučaju ne pokazuju kliničke znakove i simptome bolesti (čak i ne znaju da su nosioci mutacije). Međutim, ukoliko potomci naslede oba mutirana genska alela od svojih roditelja može doći do ispoljavanja bolesti. Postoji tačkasta mutacija na hromozomu 7, koja izaziva defekt na proteinu koji reguliše transport jona Cl kroz ćelijsku membranu (trcf).
Usled ovog defekta, žlezde produkuju veoma gust sekret koji dovodi do zapušenja izvodnih kanala ovih žlezda, što dovodi do oštećenja tkiva – pluća, pankreasa. Može doći do poremećaja semenih kanalića u testisima – sterilitet, poremećaja u sistemu za varenje, dijabetesa. Lečenje deteta sa cističnom fibrozom je svakodnevno – doživotno.
Autozomno recesivno nasledno oboljenje uzrokovano smanjenom količinom ili nedostatkom faktora XI, koji je komponenta sistema koagulacije krvi. Posledica je mutacije gena F11 smeštenog na hromozomu 4. Ozbiljnost kliničke slike varira. Pacijenti sa ovim poremećajem imaju problema sa zaustavljanjem krvi nakon povreda ili hiruških intervencija poput stomatoloških procedura, tonzilektomije, hirurških zahvata na urinarnom i genitalnom traktu, kao i produžena menstrualna krvarenja. Za razliku od hemofilije A i B nema krvarenja u zglobovima. Pogađa podjednako mušku i žensku populaciju.
Autozomno recesivni poremećaj usled mutacije IKBKAP gena, što znači da roditelji mogu biti nosioci samo jednog mutiranog genskog alela, ali u tom slučaju ne pokazuju kliničke znakove i simptome bolesti (čak i ne znaju da su nosioci mutacije). Međutim, ukoliko potomci naslede oba mutirana genska alela od svojih roditelja može doći do ispoljavanja bolesti. Karakteriše se neurološkim deficitima u vidu otežanog gutanja, povraćanja sa aspiracijom, obilnim znojenjem, poremećajem govora, emocionalnom nestabilnošću i odsustvom čula ukusa, osećaja za bol, toplotu usled čega se oboleli često povređuju. Ne postoji lek, polovina obolelih umire do 30 godine života.
Fankonijeva anemija tip C nastaje usled mutacije FANCC gena. Do sada je otkriveno najmanje 50 mutacija koje mogu izazvati ovaj oblik anemije.
Osnovna uloga koštane srži je produkcija novih krvnih ćelija (eritrocita, leukocita, trombocita). Čak 90% osoba sa Fankonijevom anemijom ima oslabljenu funkciju koštane srži što je uzrok smanjene produkcije svih ćelijskih elemenata krvi.
Bolest karakteriše izrazit osećaj umora, česte infekcije, poremećaj zgrušavanja krvi. Može biti izražena različita prebojenost kože , skeletni problemi (kratki prsti), poremećaji sistema za varenje, gastrointestinalne nepravilnosti, srčane mane, problemi sa vidom ili sluhom, neplodnost. Usled nekontrolisanog rasta ćelija pojedinci mogu razviti leukemiju ili druge oblike maligniteta (10-30%).
Genetski poremećaj uzrokovan oštećenjem proteina zaduženih za popravku DNK. Nastaje usled mutacije FANCG gena. Poznato je 13 gena čije mutacije uzrokuju ovo oboljenje. Oko 20% pacijenata tokom života razvije neko maligno oboljenje, najčešće akutnu mijeloidnu leukemiju (AML). 90% bolesnika razvije insuficijenciju kostne srži do 40. godine života. 60-75% bolesnika ima urođene anomalije, niskog su rasta, uočavaju se abnormalnosti kože, ruku, glave, očiju, bubrega, ušiju. Takođe je prisutan zaostatak u mentalnom razvoju, veća učestalost nastanka infekcija i krvarenja. Očekivana životna dob je oko 30 godina.
Gošeova bolest nastaje zbog mutacije GBA gena i nasleđuje se autozomno recesivno, što znači da roditelji mogu biti nosioci samo jednog mutiranog genskog alela, ali u tom slučaju ne pokazuju kliničke znakove i simptome bolesti (čak i ne znaju da su nosioci mutacije). Međutim, ukoliko potomci naslede oba mutirana genska alela od svojih roditelja može doći do ispoljavanja bolesti.
Radi se o urođenom metaboličkom poremećaju koji se karakteriše nagomilavanjem glukocerebrozida u tkivima usled nemogućnosti enzima glukocerebrozidaze da normalno vrši svoju funkciju.
Simptomi Gošeove bolesti mogu da se jave bilo kada, u detinstvu ili odraslom dobu, Vremenom se pogoršavaju i ukoliko se bolest ne leči mogu nastati dugotrajne zdravstvene komplikacije.
Kliničku sliku karakterišu: umor i osećaj slabosti, zaostajanje u rastu kod dece, česte pojave modrica, krvarenja iz nosa ili desni, uvećanje jetre i slezine, deformiteti i bolovi u kostima.
Glutarna acidemija tip 2A nastaje zbog mutacije ETFA gena i nasleđuje se autozomno recesivno, što znači da roditelji mogu biti nosioci samo jednog mutiranog genskog alela, ali u tom slučaju ne pokazuju kliničke znakove i simptome bolesti (čak i ne znaju da su nosioci mutacije). Međutim, ukoliko potomci naslede oba mutirana genska alela od svojih roditelja može doći do ispoljavanja bolesti.
Mutacija ETFA gena dovodi do nekompletne razgradnje proteina i masti, što dovodi do nakupljanja nerazgrađenih produkata metabolizma u telu i promene pH krvi (metabolička acidoza).
Glutarna acidemija obično se pojavljuje u ranom detinjstvu kao epizodna kriza u kojoj acidoza i hipoglikemija izazivaju slabost, povraćanje, promene u ponašanju. Ove metaboličke krize mogu biti izazvane uobičajenim dečijim bolestima ili stresnim situacijama. U najtežim slučajevima nastaju i fizičke nepravilnosti poput malformacija mozga, uvećanja jetre i srca, pojave cističnih promena u telu, različite malformacije bubrega i genitalija. Nekada slabost mišića može biti prvi simptom bolesti.
Nasledno metaboličko oboljenje koje nastaje usled mutacije GLDC gena i koje karakterišu abnormalno visoki nivoi aminokiseline Glicina.
Nasleđuje se autozomno recesivno, , što znači da roditelji mogu biti nosioci samo jednog mutiranog genskog alela, ali u tom slučaju ne pokazuju kliničke znakove i simptome bolesti (čak i ne znaju da su nosioci mutacije). Međutim, ukoliko potomci naslede oba mutirana genska alela od svojih roditelja može doći do ispoljavanja bolesti.
Manifestuje se u prvim danima života ili u detinjstvu u vidu encefalopatije, letargije, hipotonije, miokloničkih pokreta, epileptičkim napadima, problema sa disanjem koji mogu dovesti do smrti. Kauzalna terapija ne postoji.
Nastaje zbog nepostojanja enzima za mobilizaciju glukoze iz glikogena, zbog čega dolazi do deponovanja glikogena u jetri i drugim tkivima.
Glkogenoza tip 1A nastaje usled mutacije G6PC gena i nasleđuje se autozomno recesivno, , što znači da roditelji mogu biti nosioci samo jednog mutiranog genskog alela, ali u tom slučaju ne pokazuju kliničke znakove i simptome bolesti (čak i ne znaju da su nosioci mutacije). Međutim, ukoliko potomci naslede oba mutirana genska alela od svojih roditelja može doći do ispoljavanja bolesti.
Mutacije G6PC gena narušava normalno funkcionisanje enzima glukoza-6-fosfataze što remeti transport glukoza-6-fosfata u ćelije i njegovu razgradnju do glukoze. Glukoza-6-fosfat koji nije razgrađen do glukoze pretvara se u mast i glikogen, koji se dalje skladišti u tkiva i organe što može biti toksično za ćelije.
Javlja se u uzrastu novorođenčeta ili između 3. i 4. meseca. Tipični simptomi su: uvećana jetra, zaostajanje u rasu i razvoju, hipoglikemija, povećanje bubrega, acidoza, hiperlipidemija, hipotonija, okruglo lice “puniji” obrazi.
Nastaje zbog nepostojanja enzima za mobilizaciju glukoze iz glikogena, zbog čega dolazi do deponovanja glikogena u jetri i drugim tkivima.
Glikogenoza tip 1B nastaje usled mutacije SLC37A4 gena i nasleđuje se autozomno recesivno, što znači da roditelji mogu biti nosioci samo jednog mutiranog genskog alela, ali u tom slučaju ne pokazuju kliničke znakove i simptome bolesti (čak i ne znaju da su nosioci mutacije). Međutim, ukoliko potomci naslede oba mutirana genska alela od svojih roditelja može doći do ispoljavanja bolesti.
Mutacije SLC37A4 gena narušava normalno funkcionisanje enzima glukoza-6-fosfat translokaze što remeti transport glukoza-6-fosfata u ćelije i njegovu razgradnju do glukoze. Glukoza-6-fosfat koji nije razgrađen do glukoze pretvara se u mast i glikogen, koji se dalje skladišti u tkiva i organe što može biti toksično za ćelije.
Javlja se u uzrastu novorođenčeta ili tokom 3-4 meseca. Nakupine glikogena prvenstveno oštećuju bubrege i jetru, remeti njihovu funkciju i dovodi do povećanja samih organa. Česte su hipoglikemije, acidoza, rekurentne bakterijske infekcije i nedovoljan broj belih krvnih zrnaca (neutropenija).
Urođena metabolička greška usled mutacije AGL gena na hromozomu 1p21 koji kodira amilo-1,6 glikozidazu potrebnu za razgradnju glikogena, usled čega dolazi do akumulacije glikogena u jetri i mišićima.
Nasleđuje se autozomno recesivno, što znači da roditelji mogu biti nosioci samo jednog mutiranog genskog alela, ali u tom slučaju ne pokazuju kliničke znakove i simptome bolesti (čak i ne znaju da su nosioci mutacije). Međutim, ukoliko potomci naslede oba mutirana genska alela od svojih roditelja može doći do ispoljavanja bolesti.
Ispoljava se u prvim godinama života hepatomegalijom, slabošću mišića, hipoglikemijom, hiperlipidemijom, nenapredovanjem deteta, rekurentnim infekcijama, kardiomegalijom. U odraslom dobu mogući je nastanak ciroze i karinoma jetre.
Nasledni poremećaj usled nemogućnosti razgradnje glikogena u ćelijama mišića. Nasleđuje se autozomno recesivno, što znači da roditelji mogu biti nosioci samo jednog mutiranog genskog alela, ali u tom slučaju ne pokazuju kliničke znakove i simptome bolesti (čak i ne znaju da su nosioci mutacije). Međutim, ukoliko potomci naslede oba mutirana genska alela od svojih roditelja može doći do ispoljavanja bolesti
Postoje 4 forme bolesti koje se razlikuju po vremenu ispoljavanja simptoma i vrsti simptoma. Najčešće se ispoljava u detinjstvu bolovima u mišićima, grčevima, mučninom, povraćanjem prilikom umerene fizičke aktivnosti. Oštećena mišićna vlakna mogu oštetiti bubrege (mioglobinurija) i dovesti do bubrežne insuficijencije. Moguć je razvoj hiperurikemije, žutice, hemotiličke anemije, kardiomiopatije.
Nasledno oboljenje, češće se javlja u Skandinavskim zemljama usled mutacije BCS1L gena. Nasleđuje se autozomno recesivno, što znači da roditelji mogu biti nosioci samo jednog mutiranog genskog alela, ali u tom slučaju ne pokazuju kliničke znakove i simptome bolesti (čak i ne znaju da su nosioci mutacije). Međutim, ukoliko potomci naslede oba mutirana genska alela od svojih roditelja može doći do ispoljavanja bolesti
Javlja se 1 na 47000 živorođene dece u Finskoj populaciji. Dolazi do zaostalosti u rastu i razvoju, aminoacidurije, nagomilavanja gvožđa u jetri, holestaze, laktičke acidoze i rane smrti. Preživljavanje do par meseci.
Nasledna intolerancija na fruktozu nastaje zbog mutacije ALDOB gena i nasleđuje se autozomno recesivno, što znači da roditelji mogu biti nosioci samo jednog mutiranog genskog alela, ali u tom slučaju ne pokazuju kliničke znakove i simptome bolesti (čak i ne znaju da su nosioci mutacije). Međutim, ukoliko potomci naslede oba mutirana genska alela od svojih roditelja može doći do ispoljavanja bolesti.
Kao polsledica mutacije ALDOB gena dolazi do nakupljanja fruktoze u ćelijama jetre i dovodi do njihove smrti zbod svog toksičnog dejstva.
Ispoljava se u periodu detinjstva kada se u ishranu uvedu sokovi, voće i druga hrana koja sadrži fruktozu. Tipični simptomi su: mučnina, nadutost, bol u trbuhu, proliv, povraćanje, hipoglikemija. Unos fruktoze može dovesti do oštećenja jetre i nastanka žutice ili ciroze jetre. Stalno izlaganje fruktozi može rezultirati komom i na kraju smrtnim ishodom zbog oštećenja jetre ili bubrega.
Homocistinurija tip cbIE nastaje usled mutacije MTRR gena i nasleđuje se autozomno recesivno, što znači da roditelji mogu biti nosioci samo jednog mutiranog genskog alela, ali u tom slučaju ne pokazuju kliničke znakove i simptome bolesti (čak i ne znaju da su nosioci mutacije). Međutim, ukoliko potomci naslede oba mutirana genska alela od svojih roditelja može doći do ispoljavanja bolesti.
Znaci i simptomi homocistinurije obično se razvijaju u prvoj godini života, a tipični su: anemija, intelektualni poremećaji, kratkovidost, rizik od zgrušavanja krvi, osteoporoza.
Hidroletalusni sindrom (HLS) je sindrom koji nastaje usled mutacije HYLS1 gena, a nasleđuje se autozomno recesivno, što znači da roditelji mogu biti nosioci samo jednog mutiranog genskog alela, ali u tom slučaju ne pokazuju kliničke znakove i simptome bolesti (čak i ne znaju da su nosioci mutacije). Međutim, ukoliko potomci naslede oba mutirana genska alela od svojih roditelja može doći do ispoljavanja bolesti.
Radi se o smrtonosnom sindromu koji karakterišu polidaktilija, malformacije centaralnog nervnog sistema i hidrocefalus. U 50% slučajeva pronađena su oštećenja srčanih zalistaka. Trudnoća je često praćena viškom plodove vode.
Autozomno recesivno oboljenje koje nastaje usled mutacije GNE gena. Nasleđuje se autozomno recesivno, što znači da roditelji mogu biti nosioci samo jednog mutiranog genskog alela, ali u tom slučaju ne pokazuju kliničke znakove i simptome bolesti (čak i ne znaju da su nosioci mutacije). Međutim, ukoliko potomci naslede oba mutirana genska alela od svojih roditelja može doći do ispoljavanja bolesti
Uzrokuje slabost u skeletnim mišićima koja se javlja u kasnoj adolescenciji ili ranom odraslom dobu i vremenom se pogoršava. Slabost se javlja u predelu gornjeg dela nogu, bokova, ruku i ramena. Zbog otežanog hodanja većini ljudi su potrebno invalidska kolica nakon 20 godina od pojave prvih simptoma.
Metabolički poremećaj izazvan mutacijom IVD gena i nasleđuje se autozomno recesivno, što znači da roditelji mogu biti nosioci samo jednog mutiranog genskog alela, ali u tom slučaju ne pokazuju kliničke znakove i simptome bolesti (čak i ne znaju da su nosioci mutacije). Međutim, ukoliko potomci naslede oba mutirana genska alela od svojih roditelja može doći do ispoljavanja bolesti. Dolazi do nakupljanja izovalerične kiseline (miris znoja). Akutni oblik se klinički manifestuje u prvih nekoliko dana života, u vidu slabe uhranjenosti, povraćanja i respiratornog distresa, zbog čega dolazi do razvoja metaboličke acidoze, hipoglikemije i hiperamonijemije. Često dolazi do supresije koštane srži. Hronični oblik se može razviti tek za nekoliko meseci ili godina.
Oboljenje, nastaje usled mutacije TMEM216 gena koja dovodi do oštećenja malog mozga. Nasleđuje se autozomno recesivno, što znači da roditelji mogu biti nosioci samo jednog mutiranog genskog alela, ali u tom slučaju ne pokazuju kliničke znakove i simptome bolesti (čak i ne znaju da su nosioci mutacije). Međutim, ukoliko potomci naslede oba mutirana genska alela od svojih roditelja može doći do ispoljavanja bolesti.
Znaci i simptomi se javljaju u veoma ranom uzrastu zaostatkom psihomotornog razvoja. Najčešći simptomi su ataksija, hiperpnea, sleep apnea, poremećaj pokreta očnih jabučica i jezika, hipotonija. Mogu se javiti i druge malformacije kao što je polidaktilija, rascep usne ili nepca, specifičan izgled lica, zaostatak u rastu i razvoju.
Nasledno oboljenje koje se manifestuje stvaranjem plikova, bula i rana na koži i sluzokoži usled trenja ili manjih trauma. Nastaje usled mutacije LAMC2 gena koji je bitan za kodiranje keratina ili kolagena, i dovodi do stvaranja proteina koji se ne mogu grupirati u mreže i tako ćelije kože postaju osetljive i podložne razdvajanju.
Nasleđuje se autozomno recesivno, što znači da roditelji mogu biti nosioci samo jednog mutiranog genskog alela, ali u tom slučaju ne pokazuju kliničke znakove i simptome bolesti (čak i ne znaju da su nosioci mutacije). Međutim, ukoliko potomci naslede oba mutirana genska alela od svojih roditelja može doći do ispoljavanja bolesti.
Povrede i rane na digestivnom traktu dovode do hronične neuhranjenosti i usporenog rasta. Javlja se osetljivost kože, opadanje kose, ispadanje zuba, pojava bakterijskih i gljivičnih infekcija, otežano hodanje, pojava malignih tumora, sepse.
Lamelarna ihtioza tip 1 nastaje usled mutacije TGMI gena i nasleđuje se autozomno recesivno, što znači da roditelji mogu biti nosioci samo jednog mutiranog genskog alela, ali u tom slučaju ne pokazuju kliničke znakove i simptome bolesti (čak i ne znaju da su nosioci mutacije). Međutim, ukoliko potomci naslede oba mutirana genska alela od svojih roditelja može doći do ispoljavanja bolesti.
Lamelarna Ihtioza uočava se na samom rođenju ili već do druge godine života. Koža ima izgled krljušti zbog nagomilanih rožastih naslaga, dok na mestu deskvamacije (ljuštenja) nastaje crvenilo. Kod teških oblika bolesti koža izgleda poput oklopa koji remeti osnovne životne funkcije. Promene su rasprostanjene po celom telu, nezahvaćena je samo koža pregiba. Sekrecija lojnih i znojnih žlezda je smanjena.
Nasledno autozomno recesivno oboljenje koje nastaje usled mutacije LCA5 gena. Nasleđuje se autozomno recesivno, što znači da roditelji mogu biti nosioci samo jednog mutiranog genskog alela, ali u tom slučaju ne pokazuju kliničke znakove i simptome bolesti (čak i ne znaju da su nosioci mutacije). Međutim, ukoliko potomci naslede oba mutirana genska alela od svojih roditelja može doći do ispoljavanja bolesti.
Učestalost pojave je 1:40000 živorođene dece. Karakteriše ga nistagmus, oslabljeni ili odsutni pupilarni refleksi, ozbiljno oštećenje vida i slepilo.
Leighov sindrom nastaje usled mutacije LRPPRC gena i nasleđuje se autozomno recesivno, što znači da roditelji mogu biti nosioci samo jednog mutiranog genskog alela, ali u tom slučaju ne pokazuju kliničke znakove i simptome bolesti (čak i ne znaju da su nosioci mutacije). Međutim, ukoliko potomci naslede oba mutirana genska alela od svojih roditelja može doći do ispoljavanja bolesti.
Leighov sindrom karakterišu teški neurološki poremećaji usled pisutnih lezija u mozgu, koje su posledica nedostatka citohrom C oksidaze. Ispoljava se zastojem u razvoju, hipotonijom, dizmorfizmom lica . Metabolička acidoza i koma su često uzrok smrtnog ishoda.
Leukoencefalopatija nastaje usled mutacije EIF2B5 gena i nasleđuje se autozomno recesivno, što znači da roditelji mogu biti nosioci samo jednog mutiranog genskog alela, ali u tom slučaju ne pokazuju kliničke znakove i simptome bolesti (čak i ne znaju da su nosioci mutacije). Međutim, ukoliko potomci naslede oba mutirana genska alela od svojih roditelja može doći do ispoljavanja bolesti.
Leukoencefalopatija je progresivni poremećaj koji prvenstveno zahvata centralni nervni sistem (mozak i kičmenu moždinu). Naročito je zazvaćena bela masa u kojoj se nalaze nervna vlakna prekrivena mijelinskim omotačem.
Na samom rođenju nisu prisutni znakovi i simptomi bolesti. Kod dece je često usporen motorni razvoj, a zatim se kasnije javljaju spazam mišića i poteškoće u koordinaciji pokreta. Može doći i do mentalnog propadanja. Kod ženskih osoba patološkim procesom mogu biti zahvaćeni i jajnici (disgeneza jajnika). Stres, infekcije ili povrede mogu provocirati napade i pogoršanje bolesti, koji se smenjuju sa periodima stabilnosti.
Nasledno recesivno oboljenje. Učestalost pojave 1:100000 muškaraca. Uzrokovano je genetskom mutacijom LHCGR gena. Lajdigove ćelije u testisima izlučuju polne hormone koji su važni za normalan seksualni razvoj. Poremećaj dovodi do niza genitalnih abnormalnosti (mikropenis, hipospadija, bifidni skrotum, kriptorhizam). Kod ljudi sa težim oblikom poremećaja izostaje razvoj sekundarnih polnih karakteristika, ili nastaje razvoj ženskih polnih karakteristika.
Postoji više tipova oboljenja koji se razlikuju u vremenu početka ispoljavanja i težini kliničke slike. Tip 2 je najteži oblik, nastaje usled mutacije SGCB gena gena i nasleđuje se autozomno recesivno, što znači da roditelji mogu biti nosioci samo jednog mutiranog genskog alela, ali u tom slučaju ne pokazuju kliničke znakove i simptome bolesti (čak i ne znaju da su nosioci mutacije). Međutim, ukoliko potomci naslede oba mutirana genska alela od svojih roditelja može doći do ispoljavanja bolesti.
Karakteriše je progresivna miopatija koja zahvata proksimalnu muskulaturu (mišići ramenog pojasa, gornjih ekstremiteta, karlice i proksimalni delovi donjih ekstremiteta). Slabost mišića utiče na njihovu funkciju, kao i na stav tela (skolioza, izražena lordoza). Kod nekih pacijenata se javlja slabost srčanog mišića (kardiomiopatija), može se javiti i slabost respiratornih mišića gde je u težim slučajevima neophodna asistirana ventilacija.
Oboljenje, nastaje usled mutacije DLD gena gena i nasleđuje se autozomno recesivno, što znači da roditelji mogu biti nosioci samo jednog mutiranog genskog alela, ali u tom slučaju ne pokazuju kliničke znakove i simptome bolesti (čak i ne znaju da su nosioci mutacije). Međutim, ukoliko potomci naslede oba mutirana genska alela od svojih roditelja može doći do ispoljavanja bolesti.
Poremećena je razgradnja ketoanaloga: leucina, izoleucina, valina. Zbog toga se pomenute aminokiseline nagomilavaju u krvi i povišeno izlučuju u urinu. Kliničke manifestacije se ispoljavaju u prvim nedeljama života: slabo uzimanje hrane, povraćanje, tahipnea, depresija CNSa, hipotonija koja se smenjuje sa hipertonijom, konvulzije. Urin karakteristično miriše na javorov sirup.
Poremećaj uzrokovan odsustvom lipoprotein lipaze, ili deficitom njenog kofaktora APOCII, usled mutacije LPL gena. Nasleđuje se autozomno recesivno, što znači da roditelji mogu biti nosioci samo jednog mutiranog genskog alela, ali u tom slučaju ne pokazuju kliničke znakove i simptome bolesti (čak i ne znaju da su nosioci mutacije). Međutim, ukoliko potomci naslede oba mutirana genska alela od svojih roditelja može doći do ispoljavanja bolesti.
Ispoljava se visokom postprandijalnom hipertrigliceridemijom i hiperholesterolemijom. Karakterišu je ksantomi, hepatosplenomegalija, napadi pankreatitisa.
Oboljenje koje nastaje mutacijom HADHA gena, zbog čega je sprečeno pretvaranje masti u energiju. Nasleđuje se autozomno recesivno, što znači da roditelji mogu biti nosioci samo jednog mutiranog genskog alela, ali u tom slučaju ne pokazuju kliničke znakove i simptome bolesti (čak i ne znaju da su nosioci mutacije). Međutim, ukoliko potomci naslede oba mutirana genska alela od svojih roditelja može doći do ispoljavanja bolesti.
Simptomi se javljaju tokom ranog detinjstva, a to su poteškoće u hranjenju, nizak nivo energije, hipoglikemija, hipotonija, problemi sa jetrom. Kasnije u detinjstvu dolazi do razvoja perifernih neuropatija, poteškoća u disanju, ozbiljnih srčanih problema.
Intolerancija lizina nastaje usled mutacije SLC7A7 gena i nasleđuje se autozomno recesivno, , što znači da roditelji mogu biti nosioci samo jednog mutiranog genskog alela, ali u tom slučaju ne pokazuju kliničke znakove i simptome bolesti (čak i ne znaju da su nosioci mutacije). Međutim, ukoliko potomci naslede oba mutirana genska alela od svojih roditelja može doći do ispoljavanja bolesti.
Usled mutacije SLC7A7 gena poremećen je transport lizina, arginina i ornitina, čiji deficit ometa mnoge vitalne funcije. Naročito je značajno nagomilavanje amonijaka u krvi usled nedostatka arginina i ornitina, dok nedovoljna količina lizina remeti strukturu kolagena. Posledica je slabost vezivnog tkiva kao što su koža, tetive i ligament, a osteoporoza je česta pojava.
Nastaje usled mutacije BCKDHB gena i nasleđuje se autozomno recesivno , što znači da roditelji mogu biti nosioci samo jednog mutiranog genskog alela, ali u tom slučaju ne pokazuju kliničke znakove i simptome bolesti (čak i ne znaju da su nosioci mutacije). Međutim, ukoliko potomci naslede oba mutirana genska alela od svojih roditelja može doći do ispoljavanja bolesti.
Kliničke manifestacije se ispoljavaju u prvim nedeljama života: slabo uzimanje hrane, povraćanje, tahipnea, depresija CNSa, hipotonija koja se smenjuje sa hipertonijom, konvulzije. Urin karakteristično miriše na javorov sirup.
Oboljenje uzrokovano mutacijom MMAA gena i nasleđuje se autozomno recesivno, što znači da roditelji mogu biti nosioci samo jednog mutiranog genskog alela, ali u tom slučaju ne pokazuju kliničke znakove i simptome bolesti (čak i ne znaju da su nosioci mutacije). Međutim, ukoliko potomci naslede oba mutirana genska alela od svojih roditelja može doći do ispoljavanja bolesti.
Simptomi i znaci se obično ispoljavaju u ranom detinjstvu i variraju od blagih do životno ugrožavajućih. Klinička slika obuhvata povraćanje, dehidrataciju, hipotoniju, usporeno napredovanje, letargiju, hepatomegaliju, hronični pankreatitis, hroničnu bolest bubrega. Neprepoznata i nelečena bolest može dovesti do kome i smrti.
Metilmalonska acidurija tip Mut(O) nastaje usled mutacije MUT gena i nasleđuje se autozomno recesivno, , što znači da roditelji mogu biti nosioci samo jednog mutiranog genskog alela, ali u tom slučaju ne pokazuju kliničke znakove i simptome bolesti (čak i ne znaju da su nosioci mutacije). Međutim, ukoliko potomci naslede oba mutirana genska alela od svojih roditelja može doći do ispoljavanja bolesti.
Usled nedostatka enzima metilmalonil-CoA mutaze protieini i lipiti se adekvatno ne razgrađuju, što dovodi do njihovog nakupljanja u tkivima i organima . Ovo je ujedno i najteži tip metilmalonske acidurije.
Javlja se vrlo rano, već u periodu novorođenčeta. Glavni simptomi su povraćanje, dehidratacija, slab mišićni tonus, zastoj u rastu i razvoju, uvećena jetra. Često bolest zahvata bubrege i pankreas dovodeći do kome i smrtnog ishoda.
Metilmalonska acidurija i homocistinurija nastaje usled mutacije MMACHC gena i nasleđuje se autozomno recesivno, što znači da roditelji mogu biti nosioci samo jednog mutiranog genskog alela, ali u tom slučaju ne pokazuju kliničke znakove i simptome bolesti (čak i ne znaju da su nosioci mutacije). Međutim, ukoliko potomci naslede oba mutirana genska alela od svojih roditelja može doći do ispoljavanja bolesti
Usled mutacije gena nastaje poremećaj transporta vitamina B12, što ometa sintezu enzima za razgradnju proteina, mast i holesterola.
Glavne karakteristike poremećaja su zastoj u rastu i razvoju, oštećenja oka, neurološki problemi i anemije.
Metilmalonska acidurija i homocistinurija nastaje usled mutacije MMADHC gena i nasleđuje se autozomno recesivno, , što znači da roditelji mogu biti nosioci samo jednog mutiranog genskog alela, ali u tom slučaju ne pokazuju kliničke znakove i simptome bolesti (čak i ne znaju da su nosioci mutacije). Međutim, ukoliko potomci naslede oba mutirana genska alela od svojih roditelja može doći do ispoljavanja bolesti.
Usled mutacije gena nastaje poremećaj transporta vitamina B12 što ometa sintezu enzima metilmalonil-CoA mutaze i metionin sintetaze. Kao rezultat, određene aminokiseline, lipidi, holesterol se ne razgrađuju i i homocistein se ne pretvara u metionin, tako da se nakupljaju u tkiva i organe gde deluju toksično.
Simptomi se javljaju vrlo rano. Karakteristike su usporen rast i razvoj deteta, neurološki problemi, slab mišićni tonus. Većina novorođenčadi ima malu veličinu glave (mikrocefalija) i poremećaj intelektualnog razvoja. Manje uobičajene karakteristike su očni problemi i anemija. Ukoliko se ne prepozna i leči opasna je po život deteta.
Mukopolisaharidoza tip II nastaje usled mutacije IDS gena, odgovornog za sintezu enzima koji je neophodan za razgradnju složenih molekula šećera gilkozaminoglikana. U slučaju nedostatka aktivnosti enzima gikozaminoglikani se nakupljaju u lizozomima ćelija i mogu remetiti funkcije drugih proteina i pravilno funkcionisanje ćelija. .
Na samom rođenju nisu izraženi simpomi bolesti, već se javljaju između 2. i 4. godine. Mogu se uočiti pune usne, širok nos, “okrugli” obrazi, uvećan jezik. Glasne žice su voluminoznije, što rezultira dubokim i hrapavim glasom. Sužavanje disajnih puteva uzrokuje česte infekcije i kratke prekide disanja tokom sna. Često su uvećani i drugi oragni poput slezine, jetre, srca. Hidrocefalus, gubitak sluha, poremećaji vida, sindrom Karpalnog tunela i spinalna stenoza nisu redak nalaz. Nakon pete godine života rast dece se usporava, a duge kosti postaju zadebljane.
Mukopolisaharidoza tip IIIC nastaje usled mutacije HGSNAT gena, odgovornog za sinetezu enzima N-acetiltransferaze koji se nalazi u lizomomima i odgovoran je za razgradnju složenih molekula glikozaminoglikana. Nakupljanje glikozaminoglikana mogu remetiti funkcije drugih proteina i pravilno funkcionisanje ćelija. Nasleđuje se autozomno recesivno, što znači da roditelji mogu biti nosioci samo jednog mutiranog genskog alela, ali u tom slučaju ne pokazuju kliničke znakove i simptome bolesti (čak i ne znaju da su nosioci mutacije). Međutim, ukoliko potomci naslede oba mutirana genska alela od svojih roditelja može doći do ispoljavanja bolesti.
Simtomi se javljaju rano tokom detinjstva. Deca često imaju probleme sa govorom i ponašanjem, nemirni su, agresivni i destruktivni, narušena je socijalna interakcija. Komunikacija je otežana, česti su poremećaji spavanja i ograničene intelektualne sposobnosti. Stečene veštine se postepeno gube (razvojna regresija). Fizičke karakteristike i zahvaćenost organa je manje izražena nego kod ostalih oblika mukopolisaharidoze.
Oboljenje koje nastaje usled mutacije SUMF1 gena i nasleđuje se autozomno recesivno, što znači da roditelji mogu biti nosioci samo jednog mutiranog genskog alela, ali u tom slučaju ne pokazuju kliničke znakove i simptome bolesti (čak i ne znaju da su nosioci mutacije). Međutim, ukoliko potomci naslede oba mutirana genska alela od svojih roditelja može doći do ispoljavanja bolesti.
Javlja se između prve i druge godine uzrasta. Simptomi su oštećenje sluha, oštećenje skeletnog sistema, hepatosplenomegalija, ihtioza, usporen rast i razvoj. Bolest je fatalna sa simptomima koji uključuju neurološka oštećenja i ozbiljnu mentalnu retardaciju.
Miotubularna miopatija nastaje usled mutacije MTM1 gena i nasleđuje se preko X hromozoma.
Osobe sa ovim stanjem imaju slabost skeletnih mišića i smanjen tonus mišića (hipotonija) koji su obično vidljivi pri rođenu. Hranjenje može biti otežano, kao i sedenje, stajanje, hodanje. Pojedina deca nemaju adekvatnu mišićnu snagu da samostalno dišu već koriste mehaničku potporu (mehanička ventilacija). Mišići oka i lica su takođe zahvaćeni što dovodi do oftalmoplegije. Refleksi mogu biti odsutni. Zbog izrazite slabosti mišića javlja se zakrivljenost kičmenog stuba (skolioza), zglobne deformacije kukova i kolena. Deca sa miotubularnom miopatijom mogu imati povećan obim glave, usko lice i nepce koje je visoko postavljeno, a takođe i česte respiratorne tegobe.
Nasledno oboljenje uzrokovano mutcijom MPV17 gena, usled čega je poremećeno stvaranje proteina koji ulazi u sastav unutrašnje membrane mitohondrija i važan je za očuvanje mitohondrijalne DNK.
Nasleđuje se autozomno recesivno, što znači da roditelji mogu biti nosioci samo jednog mutiranog genskog alela, ali u tom slučaju ne pokazuju kliničke znakove i simptome bolesti (čak i ne znaju da su nosioci mutacije). Međutim, ukoliko potomci naslede oba mutirana genska alela od svojih roditelja može doći do ispoljavanja bolesti.
Funkcija mitohondrija u mnogim tkivima je oštećena naročito u mozgu i jetri. Ispoljava se u ranom detinjstvu povraćanjem, dijarejom, usporenim napredovanjem, hipoglikemijom, laktat acidozom, hepatomegalijom, holestazom, retko i kancerom jetre. Neurološki problemi poput usporenog razvoja, hipotonije, periferne neuropatije dovode do smrti u prvim godinama života.
Kasni infantilni oblik neuronalne ceroidne lipofuscinoze nastaje usled mutacije CLN8 gena i naslađuje se autozomno recesivno, što znači da roditelji mogu biti nosioci samo jednog mutiranog genskog alela, ali u tom slučaju ne pokazuju kliničke znakove i simptome bolesti (čak i ne znaju da su nosioci mutacije). Međutim, ukoliko potomci naslede oba mutirana genska alela od svojih roditelja može doći do ispoljavanja bolesti.
Neuronalna ceroidna lipofuscinoza predstavlje grupu poremećaja koji se zajednički mogu nazvati Batenova bolest. Mutacija CLN8 gena uzrokuje smanjenu sintezu lipida u moždanim ćelijama, dok u težim oblicima bolesti dolazi do nakupljana proteina u lizozomima ćelija celog tela što rezultira njihovim propadanjem. Nervne ćelije su posebno ranjive i njihova oštećenja dovode do neuroloških znakova i simptoma bolesti. Blaže oblike bolesti karakterišu učestali napadi epilepsije i pad intelektualnih sposobnosti u periodu od 5. do 10. godine. Generalizovane kloničko-tonične napade karakterišu konvulzije, krutost mišića i gubitak svesti. Prisuni su poremećaji koordinaciji i ravnoteže. Uobičajen je gubitak vida. Zivotni vek je skraćen.
Bolest nervnog sistema, koja počinje u detinjstvu. Osnovni uzrok je mutacija MFSD8 gena, koja izaziva nedostatak enzima koji je odgovoran za izbacivanje otpada iz ćelija centralnog nervnog sistema. . Nasleđuje se autozomno recesivno, što znači da roditelji mogu biti nosioci samo jednog mutiranog genskog alela, ali u tom slučaju ne pokazuju kliničke znakove i simptome bolesti (čak i ne znaju da su nosioci mutacije). Međutim, ukoliko potomci naslede oba mutirana genska alela od svojih roditelja može doći do ispoljavanja bolesti.
Deca sa Batenovom bolešću pate od epileptičnih napada, progresivno gube motorne funkcije, vid i mentalne sposobnosti, da bi konačno postala potpuno slepa, vezana za krevet i bez sposobnosti komunikacije. Do danas, Batenova bolest je uvek fatalna.
Kasni infantilni oblik neuronalne ceroidne lipofuscinoze nastaje usled mutacije TPP1 gena i nasleđuje se autozomno recesivno, što znači da roditelji mogu biti nosioci samo jednog mutiranog genskog alela, ali u tom slučaju ne pokazuju kliničke znakove i simptome bolesti (čak i ne znaju da su nosioci mutacije). Međutim, ukoliko potomci naslede oba mutirana genska alela od svojih roditelja može doći do ispoljavanja bolesti.
Neuronalna ceroidna lipofuscinoza predstavlja grupu poremećaja koji se zajednički mogu nazvati Batenova bolest. Mutacija TPP1 gena narušava motorni i mentalni razvoj, a prvi simptomi pojave se već imeđu 2. i 4. godine u vidu epileptičnih napada , dok je koordinacija pokreta narušena. U najmlađem uzrastu otežano je hodanje, sedenje i govorni razvoj, zapaženo je trzanje mišića i poremećaji vida. Retko se prvi simptomi bolesti jave nakon 4. godine i tada su kliničke karakteristike blaze. Životni vek je skraćen.
Oboljenje je uzrokovano je mutacijom NBN gena i nasleđuje se autozomno recesivno, što znači da roditelji mogu biti nosioci samo jednog mutiranog genskog alela, ali u tom slučaju ne pokazuju kliničke znakove i simptome bolesti (čak i ne znaju da su nosioci mutacije). Međutim, ukoliko potomci naslede oba mutirana genska alela od svojih roditelja može doći do ispoljavanja bolesti.
Klinička slika obuhvata mikrocefaliju (bird-like face), nizak rast, imunodeficijenciju (česte rekurentne infekcije), fotosenzitivnost i sklonost ka malignitetima limfnog sistema (50% do 9-te godine). Smrt nastaje u ranom dobu usled infekcije ili maligniteta- retko dožive period adolescencije.
Teška kombinovana imunodeficijencija nastaje usled mutacije RAG2 gena i nasleđuje se autozomno recesivno, , što znači da roditelji mogu biti nosioci samo jednog mutiranog genskog alela, ali u tom slučaju ne pokazuju kliničke znakove i simptome bolesti (čak i ne znaju da su nosioci mutacije). Međutim, ukoliko potomci naslede oba mutirana genska alela od svojih roditelja može doći do ispoljavanja bolesti.
Mutacija RAG2 gena izaziva poremećaj imunog sistema koji se na neadekvatan način bori protiv bakterija, virusa i gljivica ili napada sopstvena tkiva i organe. U najmlađem periodu nastaju teške i po život opasne infekcije poput pneumonije. Česte su hronične dijareje izazvane uzročnicima koji čine fiziološku floru organizma i kod zdravih osoba ne izazivaju bolest. Sindrom je često fatalan u periodu odojčeta.
Deficit ornitin aminotransferase nastaje usled mutacije OAT gena i nasleđuje se autozomno recesivno, što znači da roditelji mogu biti nosioci samo jednog mutiranog genskog alela, ali u tom slučaju ne pokazuju kliničke znakove i simptome bolesti (čak i ne znaju da su nosioci mutacije). Međutim, ukoliko potomci naslede oba mutirana genska alela od svojih roditelja može doći do ispoljavanja bolesti.
Ornitin aminotransferaza je enzim koji je aktivan u mitohondrijama, gde pomaže razgradnju ornitina. Ornitin je uključen u ciklus uree i zadužen je za obradu viška azota (u oliku amonijaka) koji nastaje u metaboličkim procesima. Nakupljanje ornitina u krvi dovodi do gubitka vida i smanjene sinteze kreatina koji je bitan u pružanju energije za kontrakciju mišića.
Progresivni gubitak vida počinje već u ranom periodu detinjstva. Prvi simptom je kratkovidost, zatim slabiji vid u večernjim satima (noćno slepilo), gubitak perifernog vida. Katarakta je čest nalaz.
Osim toga prisutne su poteškoće prilikom hranjenja, povraćanje, koma usled visokih koncentracija ornitina u krvi.
Nasledni poremećaj ciklusa uree, procesa u kome se toksični amonijak (NH3) pretvara u ureu, u obliku koje se izbacuje iz organizma putem urina. Kao posledica se javlja povišenje koncentracije NH3 u krvi, koji ispoljava toksične efekte prvenstveno na CNS-u.
Nasleđuje se autozomno recesivno, što znači da roditelji mogu biti nosioci samo jednog mutiranog genskog alela, ali u tom slučaju ne pokazuju kliničke znakove i simptome bolesti (čak i ne znaju da su nosioci mutacije). Međutim, ukoliko potomci naslede oba mutirana genska alela od svojih roditelja može doći do ispoljavanja bolesti.
Kod novorođenčadi se ispoljava dramatično odbijanje hrane, povraćanje, tahipnea, letargija do kome. Tokom detinjstva i u adolescenciji mogu se javiti periodično povraćanje, ataksija, mentalna konfuzija, uznemirenost, agresivnost, nejasan govor, koma.
Pendred sindrom nastaje usled mutacije gena SLC26A4 i nasleđuje se autozomno recesivno, što znači da roditelji mogu biti nosioci samo jednog mutiranog genskog alela, ali u tom slučaju ne pokazuju kliničke znakove i simptome bolesti (čak i ne znaju da su nosioci mutacije). Međutim, ukoliko potomci naslede oba mutirana genska alela od svojih roditelja može doći do ispoljavanja bolesti.
Usled mutacije SLC26A4 gena, otežan je transport jona kroz ćelijsku membranu, a naročito su osetljive ćelije štitne žlezde i unutrašnjeg uha, dok u manjoj meri i ćelije bubrega, jetre i disajnih puteva. Glavni klinički znaci su: povećenje štitne žlezde, gubitak sluha i poremećaj ravnoteže usled disfunkcije vestibularnog sistema i prisutni su u periodu detinjstva.
Oboljenje koje nastaje mutacijom PEX1 gena. Nasleđuje se autozomno recesivno, što znači da roditelji mogu biti nosioci samo jednog mutiranog genskog alela, ali u tom slučaju ne pokazuju kliničke znakove i simptome bolesti (čak i ne znaju da su nosioci mutacije). Međutim, ukoliko potomci naslede oba mutirana genska alela od svojih roditelja može doći do ispoljavanja bolesti.
Ima tešku kliničku sliku i postoji karakterističan lako prepoznatljiv izgled lica: visoko čelo, epikantus, hipoplazija supraorbitalne ivice i srednja linija lica. Velika fontanela je široko otorena. Mišići su izrazito hipotonični. Brojne su anomalije na očima: katarakta, glaukom, zamućenje rožnjače, pigmentna retinopatija, displazija optičkog nerva. Jetra je uvećana. Zastoj u rastu i mentalna retardacija su različitog stepena. Oštećen je sluh, postoje teškoće sa gutanjem. Konvulzije se javljaju rano. Prisutne su bubrežne anomalije (hidronefroza, renalne kortikalne mikroiste). Kauzalna terapija ne postoji.
Oboljenje koje nastaje mutacijom PEX2 gena. Nasleđuje se autozomno recesivno, što znači da roditelji mogu biti nosioci samo jednog mutiranog genskog alela, ali u tom slučaju ne pokazuju kliničke znakove i simptome bolesti (čak i ne znaju da su nosioci mutacije). Međutim, ukoliko potomci naslede oba mutirana genska alela od svojih roditelja može doći do ispoljavanja bolesti.
Ima tešku kliničku sliku, postoji karakterističan lako prepoznatljiv izgled lica: visoko čelo, epikantus, hipoplazija supraorbitalne ivice i srednja linija lica. Velika fontanela je široko otorena. Mišići su izrazito hipotonični. Brojne su anomalije na očima: katarakta, glaukom, zamućenje rožnjače, pigmentna retinopatija, displazija optičkog nerva. Jetra je uvećana. Zastoj u rastu i mentalna retardacija su različitog stepena. Oštećen je sluh, postoje teškoće sa gutanjem. Konvulzije se javljaju rano. Prisutne su bubrežne anomalije (hidronefroza, renalne kortikalne mikroiste). Kauzalna terapija ne postoji.
Oboljenje nastaje usled mutacije PAH gena. Nasleđuje se autozomno recesivno, što znači da roditelji mogu biti nosioci samo jednog mutiranog genskog alela, ali u tom slučaju ne pokazuju kliničke znakove i simptome bolesti (čak i ne znaju da su nosioci mutacije). Međutim, ukoliko potomci naslede oba mutirana genska alela od svojih roditelja može doći do ispoljavanja bolesti.
U osnovi je deficit hidroksilacije fenilalanina usled čega ne može da se konvertuje u tirozin. U jetri nedostaje ovaj enzim. Deca se rađaju zdrava, već tokom prve godine dolazi do teške mentalne retardacije IQ 30. Fenotipski su ova deca plavog tena kose i očiju. Koža je ekcemski izmenjena, i miris urina odgovara mirisu miša. Lečenje se sprovodi doživotnim ograničenjem unosa fenilalanina do količine neophodne za izgradnju sopstvenih proteina od rođenja.
Pontocerebelarna hipoplazija tip 1A nastaje usled mutacije VRK1 gena i nasleđuje se autozomno recesivno, što znači da roditelji mogu biti nosioci samo jednog mutiranog genskog alela, ali u tom slučaju ne pokazuju kliničke znakove i simptome bolesti (čak i ne znaju da su nosioci mutacije). Međutim, ukoliko potomci naslede oba mutirana genska alela od svojih roditelja može doći do ispoljavanja bolesti.
Neadekvatna razvijenost mozga je glavno obeležje ovog stanja, uslovljava odložen telesni razvoj, probleme kretanja i poremećaj intelektualnog razvoja. Pontocerebelarnu hipoplaziju prati mikrocefalija (smanjen obim glave) koja obično nije vidljiva na rođenju, ali postaje primetna već u ranom detinjstvu. U periodu novorođenčeta izraženi su poremećaji mozga, otežano je hranjenje, dok se kasnije javlja i otežano kretanje usled gubitaka motornih neurona u kičmenoj moždini. Pojedini imaju i oslabljen mišićni tonus, deformacije zglobova i otežano disanje.
Pontocerebelarna hipoplazija tip 2D nastaje usled mutacije SEPSECS gena i nasleđuje se autozomno recesivno, što znači da roditelji mogu biti nosioci samo jednog mutiranog genskog alela, ali u tom slučaju ne pokazuju kliničke znakove i simptome bolesti (čak i ne znaju da su nosioci mutacije). Međutim, ukoliko potomci naslede oba mutirana genska alela od svojih roditelja može doći do ispoljavanja bolesti.
Znakovi i simptomi su blaži nego kod ostalih oblika pontocerebelarne hipoplazije. Bolest krakteriše odložen razvoj i neadekvatna razvijenost mozga i ostalih moždanih struktura. Posledice su otežano kretanje, poremećaji hranjenja i disanja.
Pontocerebelarna hipoplazija tip 2D nastaje usled mutacije VPS53 gena i nasleđuje se autozomno recesivno, što znači da roditelji mogu biti nosioci samo jednog mutiranog genskog alela, ali u tom slučaju ne pokazuju kliničke znakove i simptome bolesti (čak i ne znaju da su nosioci mutacije). Međutim, ukoliko potomci naslede oba mutirana genska alela od svojih roditelja može doći do ispoljavanja bolesti.
Kod novorođenčadi prisutna je disfagija, otežano disanje, generalizovani klonus. Diskinezija sa mešovitom spastičnošću kao što su horeja, atetoza, diskinezija se razvijaju kasnije. Mikrocefalija, centralno oštećenje vida, zaostatak u psihomotornom razvoju, hipotonija, nemogućnost govora su takođe prisutni simptomi. Oboljenje se najčešće fatalno okončava u ranom detinjstvu.
Mutacije DNAH5 i DNAI1 odgovorne su za nastanak 30% svih slučajeva primarne cilijarne diskenezije, dok se mutacije drugih gena javljaju u malom procentu ili je uzrok nepoznat.
Primarna cilijarna diskenezija nastaje usled mutacije DNAH5 gena i nasleđuje se autozomno recesivno, , što znači da roditelji mogu biti nosioci samo jednog mutiranog genskog alela, ali u tom slučaju ne pokazuju kliničke znakove i simptome bolesti (čak i ne znaju da su nosioci mutacije). Međutim, ukoliko potomci naslede oba mutirana genska alela od svojih roditelja može doći do ispoljavanja bolesti.
DNAH5 gen odgovoran je za sintezu proteina dineina koji je neophodan za kretanje cilija. Treplje (cilije) su tanki, cilindrični, prstoliki izvrati koji polaze sa površine ćelija. Građene su po sličnom organizacionom planu kao bičevi. Njihova uloga je da svojim pokretima omoguće kretanje sluzi ili drugih supstanci preko površine različitih ćelija epitela.
Usled mutacije DNAH5 gena nastaje neadekvatan ili potpuno nedostaje protein dinein. Kao rezultat toga cilija gubi sposobnost pokreta.
Poremećaj karakterišu hronične infekcije disajnih puteva, nazalna kongestija, hronični kašalj, dok većina novorođenčadi ima otežano disanje na rođenju. Cilije u respratornom traktu imaju ulogu da svojim pokretima omoguće eliminaciju sluzi zajedno sa česticama i bakterijma, a takođe i fetusne tečnosti iz pluća. Hronične respiratorne infekcije vremenom dovode do nastanka bronhiektazija. Može se zapaziti i nepravilan položaj organa u grudima i trbuhu. U oko 12% slučajeva javljaju se abnormalnosti srca, jetre i slezine. Primarna cilijarna diskenezija takođe može dovesti do neplodnosti, ponavljajućih infekcija uha koje nose rizik od gubitka sluha
Mutacije DNAH5 i DNAI1 odgovorne su za nastanak 30% svih slučajeva primarne cilijarne diskenezije, dok se mutacije drugih gena javljaju u malom procentu ili je uzrok nepoznat.
Primarna cilijarna diskenezija nastaje usled mutacije DNAI1 gena i nasleđuje se autozomno recesivno, , što znači da roditelji mogu biti nosioci samo jednog mutiranog genskog alela, ali u tom slučaju ne pokazuju kliničke znakove i simptome bolesti (čak i ne znaju da su nosioci mutacije). Međutim, ukoliko potomci naslede oba mutirana genska alela od svojih roditelja može doći do ispoljavanja bolesti.
DNAI1 gen odgovoran je za sintezu proteina dineina koji je neophodan za kretanje cilija. Treplje (cilije) su tanki, cilindrični, prstoliki izvrati koji polaze sa površine ćelija. Građene su po sličnom organizacionom planu kao bičevi. Njihova uloga je da svojim pokretima omoguće kretanje sluzi ili drugih supstanci preko površine različitih ćelija epitela.
Usled mutacije DNAI1 gena nastaje neadekvatan ili potpuno nedostaje protein dinein. Kao rezultat toga cilija gubi sposobnost pokreta.
Poremećaj karakterišu hronične infekcije disajnih puteva, nazalna kongestija, hronični kašalj, dok većina novorođenčadi ima otežano disanje na rođenju. Cilije u respratornom traktu imaju ulogu da svojim pokretima omoguće eliminaciju sluzi zajedno sa česticama i bakterijma, a takođe i fetusne tečnosti iz pluća. Hronične respiratorne infekcije vremenom dovode do nastanka bronhiektazija. Može se zapaziti i nepravilan položaj organa u grudima i trbuhu. U oko 12% slučajeva javljaju se abnormalnosti srca, jetre i slezine. Primarna cilijarna diskenezija takođe može dovesti do neplodnosti, ponavljajućih infekcija uha koje nose rizik od gubitka sluha.
Primarna hiperoksalurija tip 3 nastaje usled mutacije HOGA1 gena i nasleđuje se autozomno recesivno, što znači da roditelji mogu biti nosioci samo jednog mutiranog genskog alela, ali u tom slučaju ne pokazuju kliničke znakove i simptome bolesti (čak i ne znaju da su nosioci mutacije). Međutim, ukoliko potomci naslede oba mutirana genska alela od svojih roditelja može doći do ispoljavanja bolesti.
Poremećaj je uzrokovan prekomernom proizvodnjom supstance zvane oksalat. Oksalat se filtrira kroz bubrege i izlučuje u visokoj koncentraciji urinom (hiperoksalurija), što vodi stvaranju bubrežnih kamenaca još u ranom detinjstvu. Tokom prolaska kroz urinarni sistem oksalat se može vezati za kalcijum pri čemu formira kalcijum oksalat, koji svojim prolaskom oštećuje bubrege i druge organe i dovodi do prisustva krvi u mokraći (hematurija). Vremenom, funkcija bubrega opada tako da više nisu u mogućnosti da izlučuju velike količine oksalata, zbog čega raste njihov nivo u krvi, a zatim se taloži u druga tkiva (sistemska oksaloza).
Nasledno autozomno recesivno oboljenje, koja nastaje usled mutacije CTSK gena koji kodira enzim Katepsin K koji je smešten u hromozomu 1.
Nasleđuje se autozomno recesivno, što znači da roditelji mogu biti nosioci samo jednog mutiranog genskog alela, ali u tom slučaju ne pokazuju kliničke znakove i simptome bolesti (čak i ne znaju da su nosioci mutacije). Međutim, ukoliko potomci naslede oba mutirana genska alela od svojih roditelja može doći do ispoljavanja bolesti.
Lizozomalno oboljenje koje se karakteriše patuljastim rastom, povećanom gustinom kostiju (osteoskleroza/osteopetroza) i krtim kostima. Druga obeležja mogu biti kifoskolioza, deformiteti trupa, gotsko nepce, kljunasti nos. Pacijenti nemaju skraćen životni vek, najčešće se javljaju zbog učestalih preloma.
Nasledno X vezano oboljenje koje nastaje mutacijom PDHB gena. Češće obolevaju muška deca. Metabolički oblik se javlja kao laktička acidoza, a neurološki oblik kao hipotonija, letargija, strukturne abnormalnosti mozga, mentalna retardacija, spasticitet, atrofija mozga.
Oboljenje nastaje usled mutacije RLBP1 gena smeštenog na hromozomu 15 koji je neophodan za normalnu funkciju čepića i štapića u oku.
Nasleđuje se autozomno recesivno, što znači da roditelji mogu biti nosioci samo jednog mutiranog genskog alela, ali u tom slučaju ne pokazuju kliničke znakove i simptome bolesti (čak i ne znaju da su nosioci mutacije). Međutim, ukoliko potomci naslede oba mutirana genska alela od svojih roditelja može doći do ispoljavanja bolesti.
Kliničke karakteristike su smanjena širina vidnog polja, noćno slepilo od ranog detinjstva.
Pigmentozni retinitis tip 25 nastaje usled mutacije EYS gena i nasleđuje se autozomno recesivno, , što znači da roditelji mogu biti nosioci samo jednog mutiranog genskog alela, ali u tom slučaju ne pokazuju kliničke znakove i simptome bolesti (čak i ne znaju da su nosioci mutacije). Međutim, ukoliko potomci naslede oba mutirana genska alela od svojih roditelja može doći do ispoljavanja bolesti.
Poremećaj karakterišu gubitak fotoreceptora, što uslovljava progresivni gubitak vida. Prvi znak je obično gubitak noćnog vida već u periodu detinjstva kada se zapaža otežano kretanje u slabije osvetljenim prostorijama. Kasnije se mogu pojaviti slepe mrlje koje se stapaju i dovode do nastanka tunelskog vida. Bolest napreduje i zahvata centralni vid zbog čega je otežano čitanje, vožnja, prepoznavanje lica. Vremenom postaju potpuno slepi.
Genetsko oboljenje očiju koje dovodi do gubitka vida koje se nastaje mutacijom gena koji kodira DHDDS enzim. Nasleđuje se autozomno recesivno, što znači da roditelji mogu biti nosioci samo jednog mutiranog genskog alela, ali u tom slučaju ne pokazuju kliničke znakove i simptome bolesti (čak i ne znaju da su nosioci mutacije). Međutim, ukoliko potomci naslede oba mutirana genska alela od svojih roditelja može doći do ispoljavanja bolesti.
Kliničke manifestacije su noćno slepilo, periferni gubitak vida, suženje vidnog polja, degeneracija retine se uočava pri pregledu očnog dna.
Nastaje zbog deficita 4 različite enzimske varijante, ali sve imaju isti rezultat – taloženje heparan sulfata.
Nasleđuje se autozomno recesivno, što znači da roditelji mogu biti nosioci samo jednog mutiranog genskog alela, ali u tom slučaju ne pokazuju kliničke znakove i simptome bolesti (čak i ne znaju da su nosioci mutacije). Međutim, ukoliko potomci naslede oba mutirana genska alela od svojih roditelja može doći do ispoljavanja bolesti.
Karakteristični su blagi somatski simptomi i teško oštećenje CNSa. Ova deca su u ranom uzrastu hiperaktivna. Krajem prve decenije života počinje brzo neurološko propadanje, nestabilan hod i vrlo brzo postaju vezani za krevet, uz razvoj mentalne retardacije. Smrt nastupa oko 15 godine života.
Česta kombinovana imunodeficijencija nastaje usled mutacije DCLRE1C gena i nasleđuje se autozomno recesivno, što znači da roditelji mogu biti nosioci samo jednog mutiranog genskog alela, ali u tom slučaju ne pokazuju kliničke znakove i simptome bolesti (čak i ne znaju da su nosioci mutacije). Međutim, ukoliko potomci naslede oba mutirana genska alela od svojih roditelja može doći do ispoljavanja bolesti.
U novorođenačkom periodu javljaju se učestale i trajne infekcije zbog oslabljenog ćelijskog i humoralnog imunog odgovora. U krvi je smanjen broj leukocita (leukopenija).
Česta kombinovana imunodeficijencija nastaje usled mutacije gena IL2RG i nasleđuje se preko X hromozoma. IL2RG gen zadužen je za sintezu proteinskog dela receptora koji su značajni za pravilno funkcionisanje imunološkog sistema.
Poremećaj karakterišu perzistentne infekcije jer im nedostaju potrebne imunološke ćelije za odbranu organizma od bakterija, virusa i gljivica. Već u periodu novorođenčeta razvijaju se hronična dijareja, gljivične infekcije i osip po koži.
Nastaje usled mutacije HBB gena i nasleđuje se autozomno recesivno, što znači da roditelji mogu biti nosioci samo jednog mutiranog genskog alela, ali u tom slučaju ne pokazuju kliničke znakove i simptome bolesti (čak i ne znaju da su nosioci mutacije). Međutim, ukoliko potomci naslede oba mutirana genska alela od svojih roditelja može doći do ispoljavanja bolesti.
Eritrociti dobijaju srpasti izgled i postaju skloni hemolizi. Zbog toga dolazi do pada koncetracije kiseonika u krvi i anemije. To dovodi do tromboze i infarkta u raznim organima – koži, slezini, plućima, kostima, bubrezima, poremećaja u cirkulaciji i nervnom sistemu, hepatomegalije, hipoksije, kardiogenog šoka i potencijalne smrti. Bolest se ne ispoljava odmah po rođenju, ima hroničnu evoluciju sa periodima bolova i hemolize. Većina bolesnika umire u mladalačkom dobu usled infekcije i insuficijencije srca i drugih organa.
Neurokutano oboljenje, koje nastaje usled mutacije ALDH3A2 gena na hromozomu 17.
Nasleđuje se autozomno recesivno, što znači da roditelji mogu biti nosioci samo jednog mutiranog genskog alela, ali u tom slučaju ne pokazuju kliničke znakove i simptome bolesti (čak i ne znaju da su nosioci mutacije). Međutim, ukoliko potomci naslede oba mutirana genska alela od svojih roditelja može doći do ispoljavanja bolesti.
To je forma ihtioze koja se uočava po rođenju. Bolest karakteriše trijada koju čini ihtioza, paraplegija i intelektualna zaostalost.
Steroid-rezistentni nefrotski sindrom nastaje usled mutacije NPHS2 gena i nasleđuje se autozomno recesivno, što znači da roditelji mogu biti nosioci samo jednog mutiranog genskog alela, ali u tom slučaju ne pokazuju kliničke znakove i simptome bolesti (čak i ne znaju da su nosioci mutacije). Međutim, ukoliko potomci naslede oba mutirana genska alela od svojih roditelja može doći do ispoljavanja bolesti.
Javlja je najčešće nakon 3. meseca ili najkasnije do 1. godine života. Bubrežna funkcija se rapidno pogoršava i razvija se hronična bubrežna insuficijencija. Osnovne kliničke manifestacije su proteinurija, hipoalbuminemija, hiperlipidemija i pojava edema. Teška hipertenzija je čest nalaz.
Stuv-Videmanov sindrom nastaje usled mutacije LIFR gena i nasleđuje se autozomno recesivno, što znači da roditelji mogu biti nosioci samo jednog mutiranog genskog alela, ali u tom slučaju ne pokazuju kliničke znakove i simptome bolesti (čak i ne znaju da su nosioci mutacije). Međutim, ukoliko potomci naslede oba mutirana genska alela od svojih roditelja može doći do ispoljavanja bolesti.
Kod novorođenčadi prisutna je disfunkcija autonomnog nervnog sistema koja dovodi do otežanog hranjenja, gutanja i disanja. Teške epizode povišene temperature praćene su pojačanim znojenjem. Poremećaj prati zakrivljenost dugih kostiju, deformacije zglobova kolena i laktova. Ostale karakteristike uključuju smanjenu gustinu kosti (osteopenija). Ishod je letalan upravo zbog poremećaja disanja i regulacije telesne temperature.
Oboljene koje nastaje usled mutacije HEXA gena i nasleđuje se autozomno recesivno, što znači da roditelji mogu biti nosioci samo jednog mutiranog genskog alela, ali u tom slučaju ne pokazuju kliničke znakove i simptome bolesti (čak i ne znaju da su nosioci mutacije). Međutim, ukoliko potomci naslede oba mutirana genska alela od svojih roditelja može doći do ispoljavanja bolesti.
Nastaje zbog deficita enzima Heksozaminidaze A, i nagomilavanja gangliozida GM2 u neuronima CNSa i perifernim ganglijama vegetativnog nervnog sistema. Nema znakova zahvaćenosti viscelarnih organa. Bolest se ispoljava u prvim mesecima života gubitkom sposobnosti fokusiranja predmeta i hiperakuzijom. U najboljem slučaju deca mogu da nauče da sede i puze, ali ne i da hodaju. Veoma malo komuniciraju sa okolinom. Dominiraju teška hipotonija (žablji položaj) i slepilo. Mogu imati makrocefaliju, konvulzije i znake decerebracije. Smrt obično nastupa između druge i četvrte godine života.
Nastaje usled mutacije PCDH15 gena i nasleđuje se autozomno recesivno, što znači da roditelji mogu biti nosioci samo jednog mutiranog genskog alela, ali u tom slučaju ne pokazuju kliničke znakove i simptome bolesti (čak i ne znaju da su nosioci mutacije). Međutim, ukoliko potomci naslede oba mutirana genska alela od svojih roditelja može doći do ispoljavanja bolesti.
Pacijenti sa Ušerovim sindromom tip 1 (USH 1) se rađaju potpuno gluvi i kasnije imaju problema i sa održavanjem ravnoteže. Noćno slepilo i gubitak perifernog vida – javljaju se u adolescenciji. Zbog problema sa održavanjem ravnoteže ova deca kasno počinju da samostalno sede i retko prohodaju pre osamnaestog meseca života. Ne postoji kauzalna terapija.
Ušerov sindrom tip 3 nastaje usled mutacije CLRN1 gena i nasleđuje se autozomno recesivno, što znači da roditelji mogu biti nosioci samo jednog mutiranog genskog alela, ali u tom slučaju ne pokazuju kliničke znakove i simptome bolesti (čak i ne znaju da su nosioci mutacije). Međutim, ukoliko potomci naslede oba mutirana genska alela od svojih roditelja može doći do ispoljavanja bolesti.
Osnovna odlika Ušerovog sindroma je teško oštećenje vida i sluha uz poremećaje ravnoteže i koordinacije pokreta. Radi se o senzorineuralnom oštećenju sluha koje se vremenom pogoršava, dok je sluh na rođenju očuvan. Gubitak sluha uočava se tokom detinjstva nakon razvoja govora i vremenom postaje sve ozbiljniji.
Gubitak vida počinje tokom perioda adolescencije, najpre pojavom slepih mrlja u perifernom vidnom polju koje se vremenom spajaju. Moguća je i pojava katarakte.
Volmanova bolest je poremećaj koji nastaje usled mutacije LIPA gena i nasleđuje se autozomno recesivno, što znači da roditelji mogu biti nosioci samo jednog mutiranog genskog alela, ali u tom slučaju ne pokazuju kliničke znakove i simptome bolesti (čak i ne znaju da su nosioci mutacije). Međutim, ukoliko potomci naslede oba mutirana genska alela od svojih roditelja može doći do ispoljavanja bolesti.
Volmanova bolest nastaje usled nedostatka enzima neophodnog za razgradnju masti što uslovljava njihovo nakupljanje u tkivima i organima. Simptomi se uočavaju ubrzo nakon rođenja, već tokom prvih nekoliko nedelja života. Poremećaj prati ozbiljni poremećaj malapsorpcije, stanje u kome creva ne uspevaju da apsorbuju potrebne hranljive materije iz hrane. Malapsorpcija izaziva uporno i često povraćanje, dijareje, neugodan miris, masnu stolicu (steatoreja), i poremećaj uhranjenosti. Često se otkriva povećanje jetre, slezine i nakupljanje slobodne tečnosti u trbušnoj duplji (ascites).
Autozomno recesivno
NASLEĐIVANJE
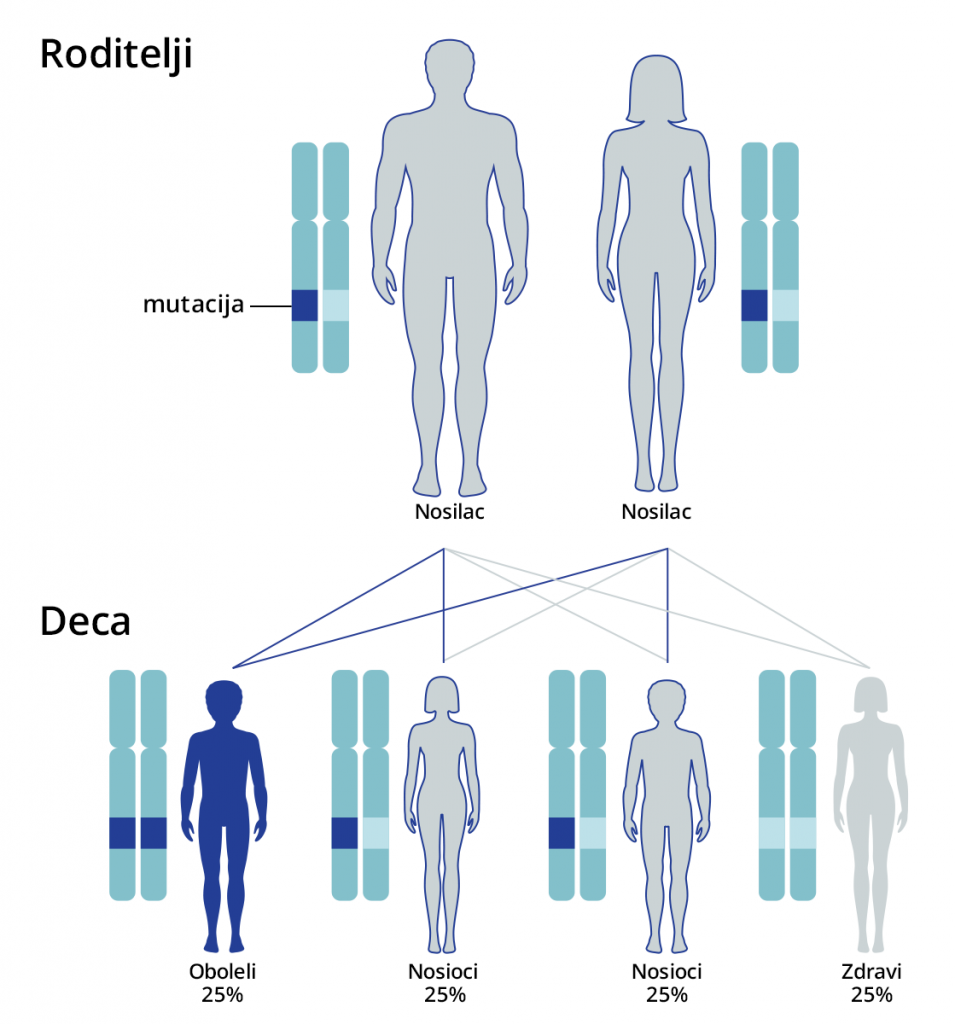
X-vezano recesivno
NASLEĐIVANJE
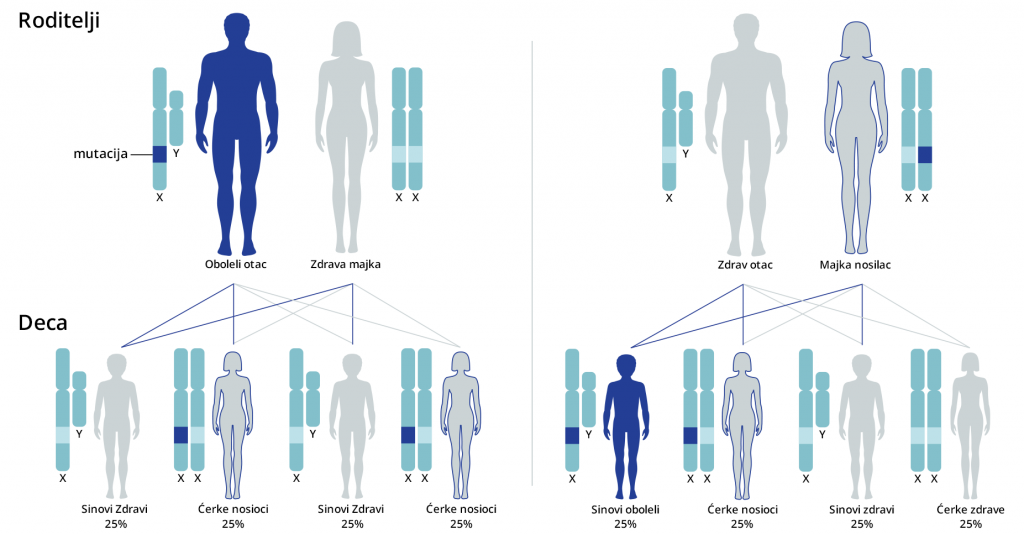
VAŽNO JE ZNATI!
Roditelji često mogu biti nosioci samo jednog mutiranog (promenjenog) genskog alela, ali u tom slučaju ne pokazuju nikakve kliničke znakove, niti simptome bolesti (čak i ne znaju da su nosioci mutacije), već promenjeni alel prenose dalje na svoje potomke.